
Submitted:
26 November 2024
Posted:
29 November 2024
Read the latest preprint version here
Preprints on COVID-19 and SARS-CoV-2
Abstract
The purpose of this communication is to clarify criticisms that have arisen in the context of our publication "Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®" (Methods Protoc. 2024, 7, 41 [1]). In the meantime, a preprint has appeared entitled "Quantification of objective concentrations of DNA impurities in mRNA vaccines" (Kaiser et al. [2]), which attempts to refute our findings. However, it does not succeed in doing so with the necessary persuasiveness. First of all, it is particularly important that Kaiser et al [2] have confirmed that our results are reproducible when the quantification is carried out using the Qubit® technology in accordance with the manufacturer's instructions, which is exactly what we did. However, it is then claimed by Kaiser et al [2] that the magnitude of the DNA impurities we have shown would be an effect of high amounts of RNA present in the samples. As a proof they quantified a defined concentration of DNA in the presence of very high concentrations of RNA with the Qubit® methodology. However, even the presence of 250 ng/µL RNA resulted only in a comparatively small increase in the DNA value of 0.655 ng/µL and this is far from explaining the DNA concentrations of 12 to 17.8 ng/µL that we have measured in several batches of Comirnaty®. Further, the preprint [2] mentioned that experiments with DNA extracted from the vaccine would show that the very low legal limits for DNA in Comirnaty® are met. However, the authors of this critique have failed to demonstrate that the extractions they performed are indeed quantitative, i.e. reflect the actual DNA contamination. However, based on the related published literature, this must be denied. With this in mind, we can finally confirm that both our methodology and our data, as published in our above-mentioned article [1], imply that DNA impurities as measured with Qubit® by us in Comirnaty® are reliable according to the manufacturer's premises for this DNA quantification technology. In this sense, the DNA values presented in the Kaiser et al. preprint [2] after extraction procedures are obviously artificial effects of the extractions performed and therefore do not represent the true DNA contamination of the concerned Comirnaty® batches.
Keywords:
mRNA vaccines
; Comirnaty®
; DNA impurities
; fluorescence spectroscopy
; Qubit® fluorometry
Introduction
In our commentary "Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®"published in Methods and Protocols (MDPI) [1], we discussed the methodological aspects of quantifying significant amounts of DNA impurities in Comirnaty®, the mRNA vaccine marketed by Pfizer and BioNTech. Recently, our results and methods as presented in this publication were publicly challenged by Kaiser et al. in a preprint article [2].
This did not come as a surprise to us, as the DNA contamination in Comirnaty® had been the subject of heated political debate prior to the publication of our peer-reviewed analysis. In the debate that followed between Rolf Marschalek, one of the authors of Kaiser et al [2] and us, Methods and Protocols and MDPI proposed a process of clarification by publishing a comment from Marschalek together with a response from us. Both sides agreed, and after some time we received the comment from Marschalek and Kaiser as his co-author, with an invitation from MDPI to respond on that. We quickly realised that we could successfully defend our publication on the basis of our own data and, surprisingly, also on the basis of data provided by Marschalek's and Kaiser's comment [unpublished]. Accordingly, we were able to refute all of Marschalek's and Kaiser's objections [unpublished] to our results and interpretations in our response submitted to Methods and Protocols on 24 September 2024 [unpublished]. In this reply we have once again demonstrated that our published data on DNA contamination in Comirnaty® [1] are scientifically sound. Both our data and our methodology are robust and the conclusion of our original publication is valid: DNA contaminations in the Comirnaty® batches we analysed were several hundred times higher than the permitted limit, providing evidence that Comirnaty® is seriously challenged in terms of drug safety.
After submitting our response, we expected that both documents, the comment by Marschalek and Kaiser and our response to it, would be reviewed by the editor and peer-reviewed according to the applicable rules [3]. However, this did not happen. Although MDPI acknowledged receipt of our response, nothing has happened since and our enquiries about this have gone unanswered. But then, about 6 weeks after we submitted our reply, Marschalek, Kaiser and further authors submitted a preprint (Kaiser et al [2]) which provides some of the criticisms as already submitted with the comment on our original publication. Obviously, Marschalek and Kaiser have decided not to continue the above-mentioned comment and response procedure and instead to try their luck with a preprint, probably due to the fact that we have backed up our data with sound evidence, as we do below also in this article.
Considerations
Comirnaty® is an mRNA vaccine targeting the SARS-CoV2 virus. It consists of a buffer solution with lipid nanoparticles containing the active ingredient, an mRNA encoding a SARS-CoV2 spike protein. A ready-to-use dose of Comirnaty® for adults consists of 300 µL containing 30 µg of mRNA [4], which means that the concentration of mRNA is 100 ng/µL Thus, any consideration of an RNA effect on DNA quantification with Qubit® concerning Comirnaty® has to focus on the effect of exactly this RNA concentration of 100 ng/µL for which particularly the manufacturer of the Qubit® system provides concrete data [5] as considered below.
Since the mRNA active ingredient of Comirnaty® is produced by transcription of specific DNA templates, the Comirnaty® production processes require large amounts of DNA as templates for in vitro synthesis of mRNA. The DNA templates used for commercial Comirnaty® are linearised plasmids from E. coli bacteria as the host. After in vitro production of the mRNA for Comirnaty®, the DNA templates have to be removed for drug safety reasons. For the commercial Comirnaty®, this was done by DNase digestion followed by filtration [4]. This means that the commercial Comirnaty® runs the risk of being contaminated with a heterogeneous mixture of DNA fragments, from small to large, as a result of the DNase digestion if the filtration doesn't remove the DNA sufficiently. That this assumption is actually true could be proven by experimental exploration of DNA lengths with the Agilent 2100 Bioanalyzer® System and the Agilent® DNA 12000 Kit as shown in Figure 1.
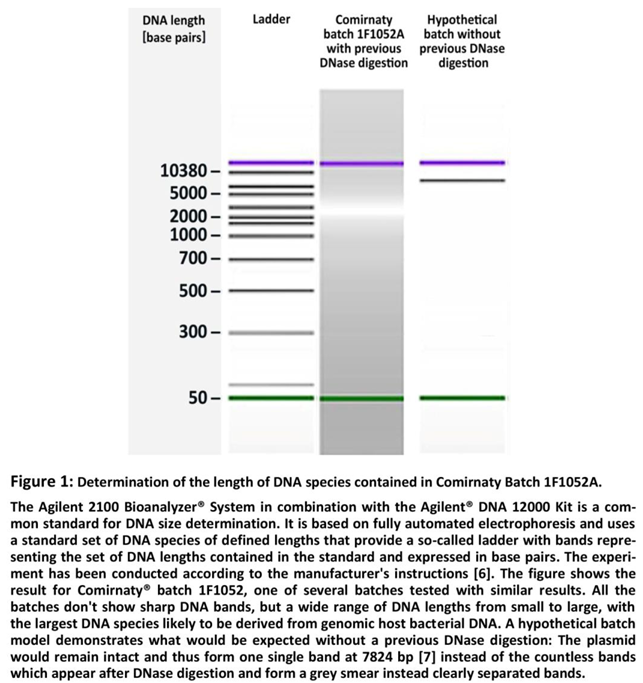
Official documents from the competent authorities indicate that the quantification of residual DNA is performed by quantitative PCR (qPCR) [7]. But this technique only measures DNA fragments containing a specific sequence of bases, and assumes that all fragments containing that sequence are of the same size [8] (→Figure 1: "Hypothetical batch without previous DNase digestion"). Since the resulting heterogeneous mixture of DNA fragments can only partially contain the qPCR target sequence. DNA fragments that do not contain this sequence escape measurement and this results in values that run the risk of reflecting only a small proportion of the actual DNA fragments, i.e. those that contain the intact qPCR target sequence. This means, that qPCR is not suitable to quantify total DNA in samples that originally contained one single uniform DNA but have been broken down by DNase digestion into a large number of DNA fragments from very short to relatively long, as it is the case with the commercialized version of Comirnaty® [7].
From this circumstance we identified the need to investigate how completely DNA fragments are actually removed from commercial Comirnaty® and how much DNA remains in marketed batches of Comirnaty®. As the DNA limit for Comirnaty® has been set by the authorities at 10 ng/dose [9,10], this value must be adhered to. Since one dose consists of 300 µL of ready-to-use diluted Comirnaty®, the final concentration of DNA contamination must be below 0.033 ng/µL to be in line with the required limit.
At this point it is important to note that Comirnaty® is a pharmaceutical drug and therefore it is important to consider whether a so called "recognized pharmaceutical rule" (this is a legal term as used e.g. in Section 55 of the German Medicines Act [11]) applies to the methods because of being mentioned in the European Pharmacopoeia. The answer can be found in the general chapter "5.14. Gene transfer medicinal products for human use" of the European Pharmacopoeia [12], which defines "gene transfer medicinal products" to include mRNA pharmaceuticals such as Comirnaty®. This chapter of the European Pharmacopoeia mentions two methods for quantifying plasmid DNA in pharmaceuticals, one based on light absorbance measurement, as used for example by the NanoDrop® device, and the other using a DNA-specific fluorescent dye and measurement of DNA-dependent fluorescence, for which several devices and methods compete, including the Qubit® device, which provides sophisticated features. This is particularly a smartly automated assay process with algorithms which take account the measurements of several light sources, filters and detectors, whereby the composition of the working solution is perfectly co-ordinated with the technical hardware and the result processing algorithms [13]. But what is more suitable for quantifying DNA in Comirnaty®?
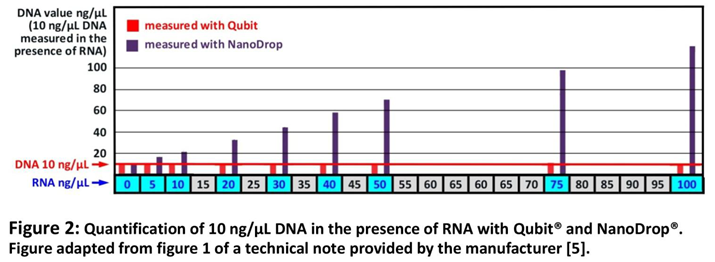
As mentioned above, due to the dosage of the active ingredient the RNA concentration in Comirnaty® is 100 ng/µL. It is therefore necessary to investigate whether this high RNA content interferes with DNA quantification. This question has been addressed by ThermoFisher/Invitrogen, the manufacturer of Qubit® and Nano-Drop®. They have provided a technical note which supports these considerations by comparing Nanodrop® (specific absorption) and Qubit® (specific fluorescence) by measuring a DNA standard of 10 ng/µL together with increasing concentrations of RNA [5]. The results are presented in a figure which shows that DNA absorption with Nanodrop® is strongly disturbed by RNA and therefore this method is disqualified under these conditions. But regarding Qubit® the manufacturer's technical note states: "In a sample containing a 10-fold excess of RNA over DNA, the concentration determined in the DNA assay was only 7% higher than the actual concentration." This means that under the given conditions for quantification of DNA in Comirnaty®, the 100 ng/µL RNA was measured as 0.7 ng/µL DNA (7% of 10 ng/µL DNA) and that this effect is fairly below the dimension of accuracy of 15% which has been defined by the manufacturer for DNA quantification with Qubit® [14].
Figure 2 shows a simplified version of the Figure published by The manufacturers ThermoFisher / Invitrogen [5] which makes it obvious that in case of the presence of high amounts of RNA, DNA quantification with Qubit® is suitable due to a low RNA effect on DNA quantification with Qubit® but absorbance measurement (Nano-Drop®) is not because the last cannot sufficiently distinguish DNA from RNA.
In general, the instructions and technical notes for Qubit® provided by the manufacturer are very clear regarding the specificity of the Qubit® DNA HS Kit in combination with the Qubit® device and show that the specificity is very high, even in the presence of contaminants such as lipids, proteins or even RNA [5,13,14]. Nevertheless, we have tested the robustness of these statements. The most appropriate way to test the influence of contaminants in a sample in which one particular substance shall be quantified is a standard addition, means the addition of known amounts of the substance to be quantified and to compare with samples without this additions. This methodology is established since decades [15] and is considered to be one of the simplest methods for detecting the influence of interferences on the result, using minimal resources and fully complies with the rules of Good Laboratory Practice (GLP) [16]. In this sense the standard addition of DNA has been applied by us regarding DNA quantification in Comirnaty® with Qubit®. According to the results of these experiments as provided in the Supplement of our original publication [1] show that DNA has been quantified accurately in our setting [17].
However, Kaiser et al. [2] claim falsely that our results would be wrong and that they actually would show an RNA effect. For this purpose, they presented data that were dimensioned in an unusual way, in that the RNA and DNA values were based on the RNA-or DNA-content of 20 or 10 µL and not expressed as concentrations, i.e. ng/µL, as given in our publication because the Qubit® device always provides the measured values this way (Figure 5). Furthermore, the x-axis of the corresponding figure was compressed so that the curve drawn by Kaiser et al. [2] is scientifically misleading. However, this can be easily corrected by converting the values provided by Kaiser et al. [2] to ng/µL and stretching the x-axis to the actual proportions. The result of this procedure is shown in Table 1 and Figure 3, where the value provided by the manufacturer's Technical Note [5] is also included.
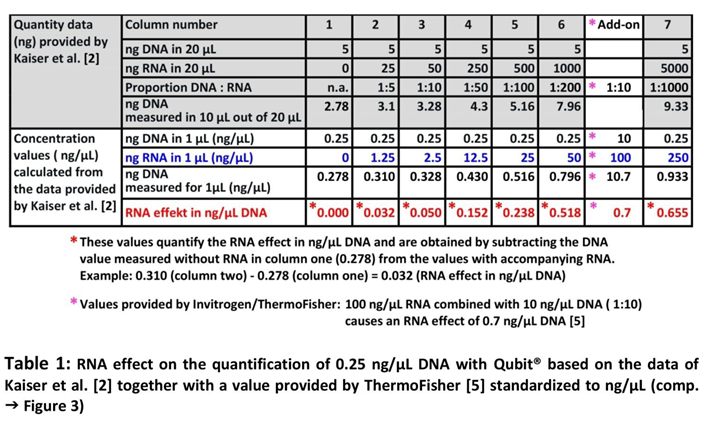
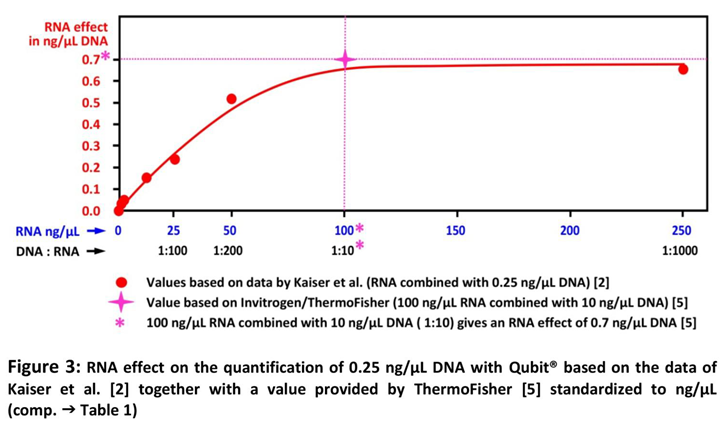
Finally the Data of Table 1 and Figure 3 demonstrate, that the RNA effect on DNA quantification with Qubit® is very small, even 250 ng/µL RNA, the 1000fold of the DNA contained in that sample (0.25 ng/µL DNA) is not higher as the RNA effect published by the manufacturer for 100 ng/µL with 0.7 ng/µL DNA if combined with 10 ng/µL DNA (10fold). Therefore, the RNA content of Comirnaty® with a dosing of 100 ng/µL RNA cannot explain the very high amounts of residual DNA as found and published by us and confirmed by Kaiser et al. [2] In particular, after dissolution of the lipid nanoparticles with Triton-X-100, the DNA values for the Comirnaty® batches investigated by us were 12 to 17.8 ng/µL and thus, even an assumed RNA effect of 1ng/µL would be far below the 15 % accuracy dimension which is claimed by the manufacturer for Qubit® DNA quantification [14]. Therefore, the assertion by Kaiser et al. [2], the values for DNA impurities in Comirnaty® batches published by us would be explainable by an RNA effect is simply false. Kaiser et al [2] themselves provided the data for this conclusion, as clearly shown in Table 1 and Figure 3 above: Surprisingly, they multiplied the original DNA value provided by the Qubit® device in ng/µL by ten to present it in their Figure 2 A [2] in a blown up way as "DNA contained in 10 µL" instead of showing the original value as expressed by the Qubit® device in ng per one µL (the other unit options ng/mL, µg/µL, µg/mL and mg/mL are not applicable, . Kaiser et al [2] did not provide, nor could we find, any scientific rationale for presenting the data at this 10-fold magnification. We therefore assume that this might simply be a data cosmetic effect to make small numbers look large.
On the other hand, when quantifying DNA in Comirnaty® using Qubit®, for samples with a low DNA concentration, e.g. 0.25 ng/µl as done by Kaiser et al. [2], and a simultaneously RNA concentration of 100 ng/µl or above, the RNA-dependent effect can exceed the accuracy limit as specified by the manufacturer with 15% deviation. This means that for a sample with such a low DNA and high RNA concentration, the RNA content of the sample would have to be reduced relative to the DNA content in order to obtain an accurate DNA value. While this sounds simple, it is challenging. In principle, a quantitative DNA extraction or a selective reduction of the RNA content would be conceivable. However, both are hampered by the fact that meaningful validation experiments would be required for such a method, which in particular can exclude an influence of the components of the Comirnaty® lipid nanoparticles. This applies in particular to the extraction of DNA, but also the reduction of RNA content using the enzyme RNase with subsequent DNA quantification using Qubit® requires a demonstration of a quantitative mode of action since the effects of the components contained in Comirnaty® on the enzymatic efficiency of RNase or the resulting RNA fragments on Qubit® measurements are unknown. A method which probably doesn't introduce interfering factors could possibly be the removal of RNA by using RNA-specific magnetic beads, a method described in the Comirnaty® EMA public assessment report [4] for the production of trail medication. However, also this method would need validation in terms of quantitativity.
In addition to the criticism of our methodology, Kaiser et al. [2] suggest in their comment a method for DNA quantification in Comirnaty® based on Phenol/Chloroform extraction, which has not been published previously in terms of quantitative extraction of DNA from pharmaceutical drugs. The same is given for another extraction used by them, the precipitation of DNA with ethanol after RNase digestion. However, Kaiser et al. [2] did that without presenting the required validation and standardization experiments. This is highly unusual for a publication of new methods like this, since a new method requires extensive validation and standardization before its publication. This is provided in international guidelines such as guidelines 7 and 11 of the Code of Conduct of the German Research Foundation [18] which applies particularly to Kaiser and Marschalek as Members of the German Goethe-University.
The DNA extraction methods suggested by Kaiser et al. are based on the extraction of DNA from Comirnaty® with phenol/chloroform respectively ethanol after RNase digestion, but both approaches are known as not being quantitative [19,20] and thus not suitable for DNA quantification in a pharmaceutical drug for human use. Of course, there are methods published how to enhance the yield of DNA e.g. from fossil bones [21] etc. when using Phenol/Chloroform, but according to our literature search there is no scientific report that this method is suitable for sample preparation for DNA quantification in pharmaceutical drugs. And so it is not surprising that Kaiser et al. [2] did not provide either experimental or literature-based evidence for a quantitative extraction of DNA using phenol/chloroform or ethanol. Further, there is no explanation why even after RNase digestion an ethanol extraction is done by them.
Such proof would require, in particular, that neither the Triton-X-100 added by Kaiser et al. [2] before the extraction nor the cationic lipids released from the lipid nanoparticles of Comirnaty® by this treatment impair the extraction procedures, which are also otherwise described by Kaiser et al. [2] only in extremely rudimentary terms. Particularly Triton-X-100 is known to interfere with phenol and ethanol, for example by formation of micelles [22,23]. Further, the cationic lipids released from the nanoparticles by the Triton extraction could interfere with the DNA in such a way that this DNA accumulates in the phenol phase or the ethanol phase and thus large DNA fractions might be discarded instead of being included in the DNA quantification. Thus, any subsequent DNA quantification to determine the DNA content of the original sample will then be compromised.
Further, it is known that phenol/chloroform marked by loss of small DNA species [19,20,21] and we have shown that the DNA contaminations in Comirnaty® consists considerably of such small DNA species (see Figure 1). Thus, Kaiser et al. [2] require to proof that the distribution pattern of DNA in terms of length has to be analyzed before the extraction takes place and then in the finally extracted DNA to show that all DNA lengths have been extracted completely. Without such a proof any DNA quantification after extraction are useless according to the applicable scientific rules.
Consequently, it has to be assumed that the very low amounts of DNA found in the extraction samples of Kaiser et al. [2] are an artificial result of the insufficiency of the extraction procedure applied by them. This has to be recognized as given until Kaiser et al. [2] provide the required proof that their extraction really works quantitatively. As long as these validation data are not provided, the extraction used by Kaiser et al. [2] doesn't allow to draw any scientific conclusions from their DNA extract - neither quantitatively nor qualitatively and completely independent from the method used for the quantification of the extracted DNA: If the quality of the sample itself is questionable, the quantification of its content is consequently questionable as well.
Thus, we have to conclude that the extraction-based methods of DNA quantification proposed by Kaiser et al. [2] is invalid for the given purpose and cannot replace the recognized pharmaceutical rule based on DNA-specific fluorescent dyes as defined by the European Pharmacopoeia and the German Medicines Act that we followed in our published [1] experiments.
A new method for the quantification of nucleic acids in mRNA vaccines was recently published by the United States National Institute of Standards and Technology (NIST) based on the quantification of RNA bases after hydrolysis using mass spectrometry (MS) [24]. This method is very promising as it does not require any extraction steps, since hydrolysis also dissolves the lipid nanoparticles of an mRNA vaccine. This is very important, as any separation steps in a purification procedure would lead to DNA losses to an undetermined extent, making any DNA measurement after these steps questionable. According to the authors, DNA impurities can be calculated from the thymine peak of the hydrolysed sample, as obtained from the same MS measurement. However, such a determination of DNA using this method is not yet ready for practical application (personal communication from NIST / Dr. Marc S. Lowenthal). Interestingly, in terms of a validation standard, NIST used a fluorescence dye-based quantification of nucleic acids after Triton treatment of the sample without any extraction of nucleic acids, as was carried out in our original publication [1] as well.
In addition to the aspects discussed above, our use of Qubit® for DNA quantification in Comirnaty® was also challenged in terms of practicality. However, from our perspective these critiques are widely based on some misunderstandings regarding the principles of the method as such. Therefore it is required to answer those questions with an explanation of essential properties of the Qubit® system which elaborates what we have already stated above. In our original Publication [1] we mentioned that technically we followed the instructions of the manufacturer, which is the standard expression for the description of well established Methods, particularly if those are suggested by the European Pharmacopoeia for the particular purpose as it is here the case.
In this light the Qubit® device is a highly automated instrument that is perfectly synchronized with DNA-specific test kits. The composition of the working solution and the algorithms for measurement and result calculation have been continuously improved over many years, as has the Qubit® device. Further, the kits contain validated DNA standards that are used to generate a validated reference curve by measuring the original standards in the Qubit® device, which automatically calculates the reference curve and checks its accuracy using special algorithms. Only when the curve has been validated in this way, the Qubit® device accepts samples for DNA quantification. In addition, the Qubit® DNA kits provide a two component working solution that contains all the components necessary for accuracy, including the fluorescent dye [25]. For a standard measurement with the Qubit® DNA HS Kit, 180 µL of the working solution is pipetted together with a sample for which the DNA content is to be quantified whereby the sample volume is 1 to 20 µL and if a volume below 20 µL is chosen, additional buffer has to be added for getting a final volume of 200 µL in the assay tube [26], as demonstrated in Figure 4.
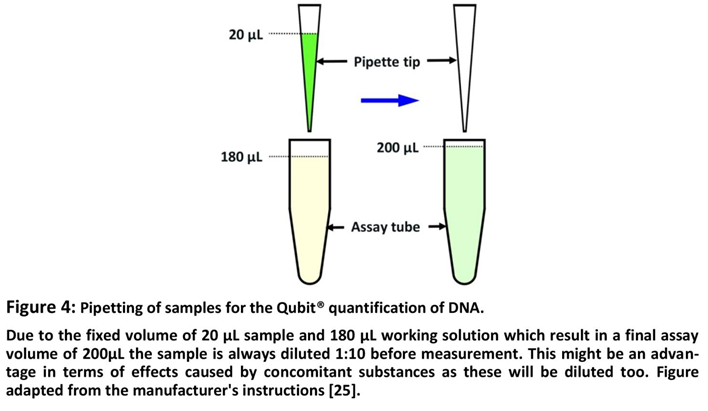
Further, for practical application according to the manufacturer's instructions the following sentence is of utmost importance: "The Qubit® dsDNA HS (High Sensitivity) Assay Kit, when used with the Qubit® Fluorometer, provides an accurate and selective method for the quantitation of sensitive DNA samples. Depending on sample volume, the assay kit is designed to be accurate for initial DNA sample concentrations of 0.005 to 120 ng/μL, providing a detection range of 0.1−120 ng." [26]
This statement differentiates between the "initial DNA sample concentration" and the "detection range". The former thus refers to the DNA concentration in the sample which can be measured with the particular assay, while the detection range indicates how much total DNA can be measured in one assay tube. Thus, if a sample with an unknown DNA concentration is analyzed with Qubit®, it is important to ensure that the test kit used is suitable in terms of its detection range. Therefore, it is generally standard practice in professional laboratories to prepare dilutions at high DNA concentrations in order to then measure at a dilution stage that lies within the intended range. If more than one dilution fulfils this requirement, all of those can be used for measurement with Qubit®. Due to the specific algorithms used automatically by the Qubit® device, the various dilutions or sample volumes lead to the same result. This is possible because the data entry mask requires to enter the volume of the original sample contained in the assay sample in µL as shown in Figure 5. Finally the output of all measurement with Qubit® is provided in the chosen unit, e.g. ng/µL DNA, as also shown in Figure 5.
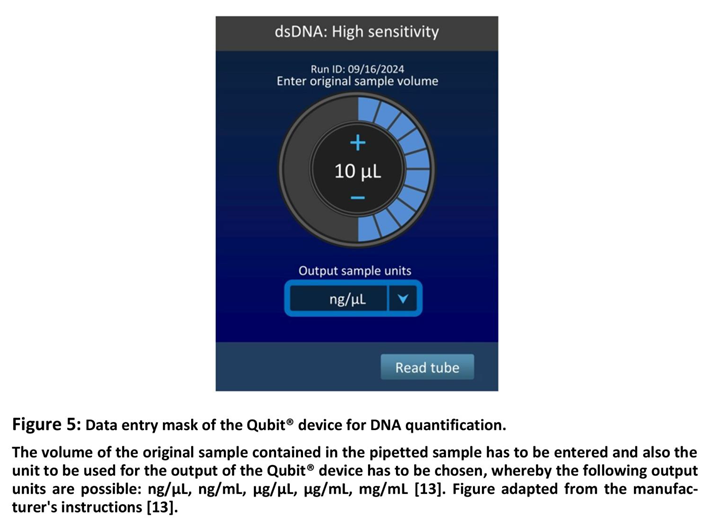
For a more detailed explanation of the Qubit® assay process, a sample with a DNA concentration of 10 ng/µL to be quantified with the Qubit® dsDNA HS Kit [25] shall serve as an example. At this value, the initial dilution series comes into play or the sample volume used must be reduced. At an initial DNA sample concentration of 10 ng/µL, an undiluted sample of 20 µL would contain 200 ng of DNA, which is considerably higher than the maximum range of 120 ng. Therefore, the sample must be diluted. This dilution can be, for example, 1:2 with then 5 ng/µL DNA, so that 20 µL of sample with then 100 ng DNA in total can be pipetted into the assay tube. However, since this sample contains only 10 µL of the original sample, this volume has to be entered into the data entry mask of the Qubit® device. Qubit's evaluation algorithm automatically includes this in the calculation of the output value, which then corresponds to the actual DNA of 10 ng/µL of the original sample, since the dilution is taken into account by the algorithm automatically.
For quality assurance purposes, it is recommended to measure several dilutions of the same original sample, which, if set up correctly, will then lead to the same result. This also applies in the case that less than 20 µL of the original sample is pipetted into the assay tube and the difference is compensated for with buffer. This is particularly important when only small sample volumes are available that are too small for a dilution series. So, as is common standard, the values given in our publication were determined with different dilutions and reproduced twice. This applies to both, DNA and RNA quantification, for which the same procedures were chosen.
Conclusions
Due to the high process quality of DNA quantification with Qubit® and the fact that even the RNA effect of 100 ng/µL RNA would lead to a DNA value of below 1 ng/µL DNA, the values we measured in several batches of Comirnaty® in the range of 12 to 17.8 ng/µL DNA are within the manufacturer's defined accuracy of 15% [14]. Therefore there is no room for a different conclusion than the following: The DNA contaminations in the Comirnaty® batches analyzed by König and Kirchner (2024) [1] are several hundred times higher than the permissible limit of 10 ng of DNA per dose or 0.033 ng/µL concentration in the ready-to-use diluted vaccine.
Author Contributions
Conceptualization, BK and JOK; methodology, BK; writing—original draft, JOK; writing—review and editing, BK and JOK; project administration, BK and JOK. All authors have read and agreed to the published version of this manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article; further inquiries can be directed to the corresponding author/s.
Acknowledgments
The authors wish to thank Irina Kouznetsova for technical and scientific support.
Conflicts of Interest
BK is CEO of Magdeburg Molecular Detection GmbH & Co. KG. The company played no role in the design of the study; the collection, analysis, or interpretation of data; the writing of the manuscript; or the decision to publish the article. J.O.K. is an author of books and articles in the context of the COVID-19 pandemic that are independent from any publisher or other third parties.
References
- König, B.; Kirchner, J.O. Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®. Methods Protoc. 2024, 7, 41. https://doi.org/10.3390/mps7030041.
- Kaiser, S. M., Kaiser, S., Reis, J., Marschalek, R., Quantification of Objective Concentrations of DNA Impurities in Mrna Vaccines. Available at SSRN:. https://doi.org/10.2139/ssrn.5009375 (accessed on 17 November 2024).
- COPE. Handling of post publication critiques. 2021, Version 1.https://publicationethics.org/sites/default/files/handling-post-publication-critiques-cope-flowchart.pdf.
- European Medicines Agency EMA. Assessment Report Comirnaty. Procedure No. EMEA/H/C/005735/0000, EMA/707383/2020 Corr.1. 19 February 2021. Available online: https://www.ema.europa.eu/en/documents/sessment-report/comirnaty-epar-public-assessment-report_en.pdf (accessed on 26 February 2024).
- Invitrogen. Comparison of Fluorescence-Based Quantitation with UV Absorbance Measurements, Technical Note. 2018. Available online: https://assets.thermofisher.com/TFS-Assets%2FLSG%2FTechnical-Notes%2Ffluorescence-UV-quantitation-comparison-tech-note.pdf (accessed on 26 February 2024).
- Agilent Technologies Inc. Kit Guide. DNA 12000 Kit for 2100 Bioanalyzer Systems. Edition: 09/2023 https://www.agilent.com/cs/library/usermanuals/public/G2938-90024_DNA7500-12000_KG.pdf (accessed on 26 February 2024).
- European Medicines Agency EMA. Rapporteur Rolling Review Critical Assessment Report, Quality Aspects COVID-19 mRNA Vaccine BioNTech. 19 November 2020. EMEA/H/C/005735/RR/xxx. Available online: https://factreview.s3.eu-central-1.amazonaws.com/uploads/2023/07/Rolling-Review-Report-Quality-COVID-19-mRNA-Vaccine-BioNTech.pdf (accessed on 16 November 2024).
- Council of Europe. Quantification and Characterisation of Residual Host-Cell DNA, Free access to Supportive Pharmacopoeial Texts in the Field of Vaccines for Human Use during the Corona virus Disease (COVID-19) Pandemic, Updated Package—October 2020, Published in Accordance with the Convention on the Elaboration of a European Pharmacopoeia (European Treaty Series No. 50). 2020: 2.6.35. Available online: https://www.edqm.eu/en/d/99080 (accessed on 26 February 2024).
- German Gouvernement. Antwort auf eine Anfrage von Abgeordneten des Deutschen Bundestages, Deutscher Bundestag Drucksache 20/9697, 20. Wahlperiode. 12 December 2023. Available online: https://dserver.bundestag.de/btd/20/096/2009697.pdf (accessed on 26 February 2024).
- German Gouvernement. Schriftliche Fragen von Abgeordneten des Deutschen Bundestages mit den in der Woche vom 11. Dezember 2023 Eingegangenen Antworten der Bundesregierung, Antwort auf Frage Nr. 104, Deutscher Bundestag Drucksache 20/9697, 20. Wahlperiode. 15 December 2023. Available online: https://dserver.bundestag.de/btd/20/098/2009807.pdf (accessed on 26 February 2024).
- German Medicines Act. Section 55. Available online: https://www.gesetze-im-internet.de/englisch_amg/englisch_amg.html#p1235 (accessed on 26 February 2024).
- Council of Europe. 5.14. Gene transfer medicinal products for human use, version legally binding until 31 March 2025, Free access to Supportive Pharmacopoeial Texts in the Field of Vaccines for Human Use during the Coronavirus Disease (COVID-19) Pandemic, Updated Package—October 2020, Published in Accordance with the Convention on the Elaboration of a European Pharmacopoeia (European Treaty Series No. 50). 2020: 2.6.35. Available online: https://www.edqm.eu/en/d/99080 (accessed on 25 June 2024).
- Invitrogen. Qubit® 4 Fluorometer, Manual. 2021. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017209_Qubit_4_Fluorometer_UG.pdf (accessed on 26 February 2024).
- Invitrogen. Comparison of Quant-iT and Qubit DNA Quantitation Assays for Accuracy and Precision, Application Note. 2016. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/Application-Notes/comparison-quantit-qubit-dna-quantitation-app-note.pdf (accessed on 26 February 2024).
- Bader, M. A systematic approach to standard addition methods in instrumental analysis. J. Chem. Educ. 1980, 57, 10, 703.
- Andersen, J. E.T. The standard addition method revisited. TrAC Trends in Analytical Chemistry. 2017, 89, 21-33. https://doi.org/10.1016/j.trac.2016.12.013.
- König, B.; Kirchner, J.O. Experiment 4. Supplemental Material for Methodological Considerations Regarding the Quantification of DNA Impurities in the COVID-19 mRNA Vaccine Comirnaty®. Methods Protoc. 2024, 7, 41. https://doi.org/10.3390/mps7030041.
- German Research Foundation. 2022. Code of Conduct. Guidelines for Safeguarding Good Research Practice. Available online: https://www.dfg.de/resource/blob/174052/1a235cb138c77e353789263b8730b1df/kodex-gwp-en-data.pdf (accessed on 12 September 2024).
- Tan, S.C.; Yiap, B.C. DNA, RNA, and protein extraction: The past and the present. J. Biomed. Biotechnol. 2009, 2009, 574398. https://doi.org/10.1155/2009/574398.
- Zdziennicka A. The adsorption properties of short chain alcohols and Triton X-100 mixtures at the water-air interface. J Colloid Interface Sci. 2009 Jul 15;335(2):175-82. https://doi.org/10.1016/j.jcis.2009.03.038. Epub 2009 Apr 2.
- Molbert, N., Ghanavi, H.R., Johansson, T. et al. An evaluation of DNA extraction methods on historical and roadkill mammalian specimen. Sci Rep 13, 13080 (2023). https://doi.org/10.1038/s41598-023-39465-z.
- N. Dharaiya, P. Bahadur. Phenol induced growth in Triton X-100 micelles: Effect of pH and phenols’ hydrophobicity, Colloids and Surfaces A: Physicochemical and Engineering Aspects. 2012. 410, 81-90. Available online: https://doi.org/10.1016/j.colsurfa.2012.06.021.
- Bergeron DE. Micellar phase boundaries under the influence of ethyl alcohol. Appl Radiat Isot. 2016 Mar;109:264-269. https://doi.org/10.1016/j.apradiso.2015.11.006. Epub 2015 Nov 5. PMID: 26585642; PMCID: PMC4937795.
- Lowenthal M.S., Antonishek A.S., Phinney K.W. Quantification of mRNA in Lipid Nanoparticles Using Mass Spectrometry. Anal. Chem. 2024, 96, 3, 1214–1222, Publication Date: 08 January 2024. Available online:https://doi.org/10.1021/acs.analchem.3c04406 (accessed on 25 June 2024).
- Invitrogen. User Manual Qubit DNA HS Assay Kits. Pub. No. MAN0017455 Rev. C, 08 Dec. 2020. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017455_Qubit_1X_dsDNA_ HS_Assay_Kit_UG.pdf (accessed on 26 February 2024).
- ThermoFisher. Invitrogen™ Qubit™ dsDNA Assay-Kits. Available online: https://www.fishersci.de/shop/products/ qubit-dsdna-hs-br-assay-kits/10616763?change_lang=true (accessed on 26 February 2024).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



