
Submitted:
27 November 2024
Posted:
28 November 2024
You are already at the latest version
Abstract
A UO22+ complex bearing N,N,N’,N’-tetraisopropyldiglycolamide (TiPDGA) and two DMF molecules was designed and prepared to explore a catalytic activities of the Lewis-acidic U centre. The cationic complex, [UO2(TiPDGA)(DMF)2]2+, was successfully obtained as a ClO4− salt under optimized reaction condition with appropriate mixing ratio between UO22+ and TiPDGA to maintain 1:1 stoichiometry, non-coordinating ClO4− counteranion to reserve coordination sites for substrate activation, and presence of extra HClO4 to suppress undesired hydrolysis of UO22+ competing with the expected complex formation. This UO22+ complex was thoroughly characterized by IR, elemental analysis, X-ray crystallography, and 1H NMR to confirm the desired [3+1+1] equatorial coordination is actually formed in the solid state, and still maintained even after dissolution in CD2Cl2. [UO2(TiPDGA)(DMF)2]2+ was further subjected to nucleophilic acyl substitution reactions of acid anhydrides to assess its activity and capability as a Lewis-acid catalyst there. Although the observed reaction rates are not very rapid, some characteristic aspects to gain reaction- and substrate-selectivity have appeared thanks to the equatorial coordination sphere sterically regulated by the tridentate auxiliary TiPDGA ligand and labile monodentate DMF molecules to activate an acid anhydride after ligand substitution.
Keywords:
Uranyl complex
; Lewis-acid catalyst
; Acyl substitution
; Reaction mechanism
1. Introduction
Although nuclear power is one of promising energy sources having overwhelmingly large energy density without CO2 emission at least during the power generation, nuclear waste issues must be resolved appropriately and urgently for sustainability of the modern and future human societies and natural environments [1]. While high level radioactive wastes (HLW) at the backend stream of the nuclear fuel cycle are most frequently focused in this connection [2,3,4,5,6], there is another issue of depleted uranium (DU) unavoidably afforded after the enrichment of 235U, the fissile, but less-abundant, isotope of U (0.72%, rest is 238U), to fabricate the nuclear fuels containing 3-5% 235U. As a result, huge amount of DU is left to be stored without any practical applications, except for a few niche uses (e.g., military purposes), despite cost and labour spent in preceded processes such as mining, refining, and conversion.
Because isotope effects in chemistry are negligibly small especially for U, the naturally occurring heaviest element, chemical applications should be convincing directions to resolve the issues of DU. Uranium has rich redox chemistry from +III to +VI [7], although lower valences such as +I and +II have also been found more recently [8,9,10,11]. While organometallic activities are usually available only in the low valent U complexes up to +IV [12,13], the most accessible valency of U is +VI to form, in most cases, UO22+ (uranyl) under the ambient condition. Indeed, functionalization of UO22+ complexes towards molecular catalytic systems have been explored extensively as reviewed previously [14,15], where either thermal or photochemical reactivity of UO22+ is employed.
UO22+ exhibits typical coordination chemistry as a hard Lewis acid [16], so that it exclusively prefers hard bases as ligands [17]. This should be the reason why it is hard to expect organometallic behaviour of UO22+ for direct activation and formation of organic bonds through oxidative additions and reductive eliminations usually observed in other complex catalysts including low valent U [12,13]. In accordance with this, we believe that one of feasible applications of UO22+ in catalytic utility is a Lewis-acid catalyst. Several successful cases such as nucleophilic addition,[18,19,20] Diels-Alder reaction [21], acyl substitution [22], and hydrosilylation [23,24] catalysed by Lewis-acidic UO22+ species have already been reported. Figure 1(a) summarizes the UO22+ catalysts employed in these former works.
Although these UO22+-based Lewis-acid catalysts actually promote aimed reactions, there are still some space to design molecular structures of them appropriately. To our best understanding, some vacancy in the equatorial plane of UO22+ must be reserved for activation of a substrate of interest. Most ordinarily, the coordination site occupied by monodentate ligand(s) such as solvent molecules (L) or OPPh3 in Figure 1(a) is offered for this purpose to avoid difficulty in preparing a coordinatively unsaturated UO22+ complex.
Considering the characteristic planar penta-coordination in the equatorial plane of UO22+, a tridentate planar ligand would be appropriate as an auxiliary scaffold to provide a regulated reaction space for the substrate activation. Due to the hardness of UO22+ described above, O-donating tridentate ligands should be preferred. For instance, N,N,N’,N’-tetraalkyldiglycolamide (TRDGA, Fig .1(b)) frequently employed in separation chemistry for trivalent lanthanides/actinides [25,26,27,28] well meets the above requirement. Furthermore, its non-charged form is expedient especially in a reaction system such as nucleophilic acyl substitution in which an acidic by-product is afforded [22].
In this study, we aimed to prepare a [3+1+1] UO22+ complex, [UO2(TRDGA)(L)2]2+ (Figure 1(b)), and to assess its catalytic activity in the nucleophilic acyl substitution of acid anhydrides as a benchmark reaction. While several UO22+-TRDGA complexes have been reported previously [26,29,30], such [3+1+1] equatorial coordination has not been attained yet. Instead, non-planar hexa-coordination with TRDGA (R = iPr, TiPDGA) and two NO3− or planar, but fully chelated, hexa-coordination by two TRDGA molecules (R = Me) were observed. To achieve the expected [UO2(TRDGA)(L)2]2+ of Figure 1, a reaction condition including selection of counteranion must be optimized.
2. Results and Discussion
2.1. Synthesis and Characterization of UO22+-Diglycolamide Complex
Yellow prismatic crystals of the desired UO22+-TiPDGA complex, [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2, were successfully obtained in 80% yield after the reaction between [UO2(DMF)5](ClO4)2 and TiPDGA from a 1:1 stoichiometric mixture of CH2Cl2 solution and recrystallization by slow diffusion of Et2O vapor to the mother liquor. This compound was thoroughly characterized by 1H NMR, IR, elemental analysis, and X-ray crystallography as summarized in Experimental section.
The molecular structure of [UO2(TiPDGA)(DMF)2]2+ is illustrated in Figure 2. A typical trans-dioxo structure of UO22+ was maintained in this complex, where the mean U≡O bond distance and the O≡U≡O angle are 1.75 Å and 179.46(19)°, respectively. This axial UO22+ is further surrounded by additional ligands such as TiPDGA and DMF molecules in its equatorial plane to form pentagonal bipyramidal coordination geometry around the U centre. All O atoms of TiPDGA are bound to U in a tridentate manner to form two 5-membered chelate rings, which may play critical roles to stabilize this complex even under presence of excess DMF molecules arising from [UO2(DMF)5]2+ [17], a source of UO22+ used in this work.
The interatomic distances from the U centre to O atoms of the amide (Oamide) and ether moieties (Oether) of TiPDGA are 2.35 Å (mean) and 2.520(3) Å, respectively, implying stronger coordination of Oamide to U compared with that of Oether in accordance with the stronger Lewis basicity of the former compared with the latter [31]. Both U−O distances between UO22+ and TiPDGA displayed in Figure 2 are significantly shorter than those found in UO2(TiPDGA)(NO3)2 reported by Kannan et al. [26] (U−Oamide = 2.40 Å (mean) and U−Oether = 2.592(3) Å). In this another UO22+-TiPDGA complex, two NO3− are involved in the equatorial coordination around UO22+. Due to the steric demands occurring in the equatorial plane of UO2(TiPDGA)(NO3)2, these NO3− exhibit different denticities; one is bidentate, while the other is monodentate, implying that there is no space wide enough to allow the bidentate coordination of both NO3−. The same trend in U−O bond distances was also found between our [UO2(TiPDGA)(DMF)2]2+ of Figure 2 and [UO2(TRDGA)2]2+ (R = Me) [29,30] showing U−Oamide = 2.42 Å (mean) and U−Oether = 2.63 Å (mean). The equatorial plane of the latter complex was somewhat expanded despite the less-bulkier methyl substituents on the amide N atoms to maintain the planar hexa-coordination around UO22+. In contrast, any steric constraints observed in the former related systems do not occur in the current [UO2(TiPDGA)(DMF)2]2+. This is because only one TiPDGA ligand is included in the coordination sphere, and also both DMF molecules are bound to U in monodentate manner (see below). This should be the reason why the shorter bond distances of U−Oamide and U−Oether most probably associated with the stronger coordination of TiPDGA has been successfully attained by employing the non-coordinating ClO4− as counteranions and stoichiometric loading of TiPDGA to UO22+ in the reaction mixture.
As displayed in Figure 2, the remaining coordination sites were occupied by two DMF molecules to saturate the equatorial plane of UO22+, where the coordination of DMF molecules to U are as strong as those of Oamide of TiPDGA as pronounced by the similar bond distances (U(1)−O(6): 2.325(4) Å, U(1)−O(7): 2.343(4) Å), which are shorter than those in [UO2(DMF)5]2+ (2.39 Å (mean) in ClO4− salt, 2.37 Å (mean) in BF4− salt) [17,32] and UO2(salophen)DMF (2.410(3) Å) [33]. This implies that the equatorial coordination sphere of [UO2(TiPDGA)(DMF)2]2+ is sterically less-constrained. Indeed, sum of equatorial bond angles around the U centre (Σeq) is 360.28° to maintain nearly ideal planarity of the typical pentagonal equatorial coordination around UO22+. Additionally, one CH2Cl2 molecule and two ClO4− are included in this crystal structure (Figure S1) as a crystalline solvent and non-coordinating counteranions of the positively charged [UO2(TiPDGA)(DMF)2]2+, respectively. Some of these isolated components were disordered even at 183 K.
In the above synthetic procedure, we have added 0.1 vol% of conc. HClO4(aq) (10 μL) to the CH2Cl2-based mother liquor (10 mL) to successfully obtain [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 of Figure 2. When this operation was skipped, another crystalline phase, [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O (Figure S2) was obtained. Although hydrolysis of UO22+ to give OH− was once considered, all isolated O atoms (O(7), O(16)) were assigned to H2O molecules because of electro-neutrality in its crystal lattice. The bond distances between U and O atoms at the apical positions were 1.76 Å (mean). The tridentate coordination of TiPDGA through amide and ether O atoms and their bond lengths (U−Oamide: 2.33 Å (mean), U−Oether: 2.526(2) Å) almost remain unchanged from those of [UO2(TiPDGA)(DMF)2]2+ shown in Figure 2. The coordination of DMF seems to be stronger than that of H2O in terms of bond lengths (U−ODMF: 2.333(3) Å vs. U−OH2O: 2.379(3) Å). Additionally, a yellow chunk material also deposited together with the crystalline [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O. Although identity of this compound has not been determined yet due to its insolubility to any organic solvents, it would be of a hydrolytic product of UO22+. This undesired reaction can be suppressed under an acidic condition, so that presence of the extra HClO4 plays a crucial role to successfully provide [UO2(TiPDGA)(DMF)2]2+ as a predominant product. For simplicity of the reaction system, we have hereafter employed the DMF solvate, [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 of Figure 2, to explore its catalytic activity in the nucleophilic acyl substitution of acid anhydrides, the main topic of this work.
The molecular structure of [UO2(TiPDGA)(DMF)2]2+ present in a CD2Cl2 solution was also studied by 1H NMR spectroscopy. The NMR signals arising from TiPDGA and DMF of this sample solution (see Experimental, Figure S3) exhibited remarkable downfield shifts compared with those of their free forms (free DMF: 7.96 (s, 2H, CHO), 2.91 (s, 3H, N(CH3)2), 2.82 (s, 3H, N(CH3)2); free TiPDGA: 4.22 (s, 4H OCH2), 3.92 (septet, 2H, CH(CH3)2), 3.45 (septet, 2H, CH(CH3)2), 1.41 (d, 12H, CH(CH3)2), 1.18 (d, 12H, CH(CH3)2)). Therefore, both TiPDGA and DMF remain coordinated to UO22+ even in the CD2Cl2 solution to maintain the original structure of [UO2(TiPDGA)(DMF)2]2+ displayed in Figure 2.
2.2. Nucleophilic Acyl Substitution Reactions Catalysed by [UO2(TiPDGA)(DMF)2]2+
In accordance with our former work [22], we have first examined the catalytic activity of [UO2(TiPDGA)(DMF)2]2+ in the reaction between acetic anhydride (Ac2O) and 2-phenylethanol (PhetOH) to afford 2-phenylethyl acetate (PhetOAc) as shown in the reaction scheme of Figure 3. Concentrations of the reactants and product at different time were determined by 1H NMR spectroscopy. Figure 3(a) illustrates typical progress of the reaction. Both reactants were congruently consumed. Along with this, production of PhetOAc was observed, and reached 90% yield at 10 h (entry 1, Table 1). Although even under absence of [UO2(TiPDGA)(DMF)2]2+ the same product was generated, its yield was only 9% at 10 h (entry 2, Table 1). Consequently, we have confirmed that this nucleophilic acyl substitution reaction was certainly catalysed by [UO2(TiPDGA)(DMF)2]2+. The reaction rate of the current system is actually slower than those reported in the former time, where the same reaction completed within 1 h to reach > 95% yield [22]. Such a difference in the kinetic aspect would be ascribed to the equatorial plane sterically regulated by TiPDGA, the auxiliary tridentate ligand employed in this work. Based on the loading ratio of [UO2(TiPDGA)(DMF)2]2+ (10 mM) with the substrates (0.5 M each) and the product yield (90% at 10 h) in entry 1 of Table 1, the turnover number (TON) of the current catalytic system was 45.
We have further surveyed capabilities of [UO2(TiPDGA)(DMF)2]2+ as a Lewis-acid catalyst in the current reaction system in use of different nucleophiles and different acid anhydrides. First, several aliphatic alcohols and phenol were tested as nucleophiles to be reacted with Ac2O (entry 3-6, Table 1). As a result, the corresponding acetate esters were generally afforded in high yields (88−100%) comparable with that of PhetOAc of entry 1 (90%), implying that a nucleophile would not largely affect the reaction mechanism.
An exception was found in use of tBuOH (entry 7, Table 1). While tBuOAc was obtained in 78% yield at 10 h as expected, isobutylene ((CH3)2C=CH2) was also generated as another product after the E1 elimination of tBuOH. Note that both of tBuOAc yield and its production rate in the current system are higher and faster than those observed in our former system catalysed by [UO2(OPPh3)4]2+ (Figure 1, 63% yield at 72 h) [22]. Furthermore, yield of isobutylene in the current system was less than 3%, which is much different from 32% yield in use of the [UO2(OPPh3)4]2+ catalyst [22]. Because H2O generated as a by-product of the E1 elimination of tBuOH readily consumes Ac2O, suppression of this side reaction is highly preferred. The steric control of the UO22+ equatorial plane with TiPDGA drawn in Figure 1 would also be favourable to prevent extra activation of bulky tBuOH for the E1 path probably through its O-coordination to the U centre of the catalyst.
In contrast to the nucleophiles, selection of acid anhydrides ((RCO)2O) has significant impact to efficiency of the current catalytic acyl substitution. As shown in entry 1 and 8-11 of Table 1, yields of the 2-phenylethyl esters of different acids remarkably decreased with an increase in length or bulkiness of R in the parent acid anhydrides. Most typically, pivalic (R = tBu) and benzoic (R = Ph) anhydrides resulted in their corresponding esters only in 10% and 6% yields, respectively. Especially for the latter case, it is hard to know whether the reaction is catalysed by the UO22+ complex or not. Even when MeOH was used as a sterically least-hindered nucleophile (entry 12, Table 1), yield of its pivaloyl ester (14%) was only slightly higher than that of the 2-phenylethyl analogue (10%, entry 10). A series of these trends implies that the acid anhydride must be activated by the [UO2(TiPDGA)(DMF)2]2+ catalyst to initiate the reaction, and that such an initial process is significantly controlled by steric effects. Details of the mechanistic aspects will be explored in the next section.
2.3. Reaction Kinetics and Catalysis Mechanism
From the data in Table 1, we have already gained some qualitative insights about mechanistic aspects of the current reaction system. As mentioned above, an acid anhydride decides yield of its product, while a nucleophile has only small impact to the reaction efficiency. Herein, more detailed and quantitative assessment of its reaction kinetics and catalysis mechanism has been done by taking the acetylation of PhetOH with Ac2O as a model reaction. Results are displayed in Figure 3(b)-(d).
When the nucleophile concentration, [PhetOH], was varied from 100 mM to 480 mM (Figure 3(b)), little differences in the reaction progress were observed unless PhetOH was consumed significantly. Indeed, the initial rate (d[PhetOAc]/dt = kini) of this series were ranging from 22.7 μM⋅s−1 to 27.5 μM⋅s−1 despite 4.8-fold variation of [PhetOH]. This indicates that the nucleophile is not involved in the rate-determining step of this catalytic reaction.
We have also examined dependency of the reaction kinetics on the concentration of the other reactant, [Ac2O]. As a result, the reaction kinetics became faster and faster with an increase in [Ac2O] (Figure 3(c)). Indeed, kini in this series linearly increased with an increase in [Ac2O]. Consequently, the rate of this catalytic reaction is first-order of [Ac2O] with a slope equal to 4.0 × 10−5 s−1 (right panel of Figure 3(c)), implying that one Ac2O molecule is involved in the rate-determining step of this reaction, most probably activation of Ac2O by the UO22+ catalyst.
Activation of Ac2O in this system would be attained through its coordination to the U centre of [UO2(TiPDGA)(DMF)2]2+. However, there would be no vacancies large enough to accept additional coordination in the equatorial plane of this UO22+ complex as displayed in Figures 2 and S4 Therefore, some of the coordinating DMF molecules should be replaced with Ac2O. If this assumption is correct, the reaction kinetics of the current catalytic system should be controlled by the concentration of DMF additionally loaded to the reaction system ([DMF]add). As a matter of fact, the reaction rate significantly decreased with an increase in [DMF]add (Figure 3(d)). Therefore, the above hypothesis was found to be correct.
To precisely assess the dependency of kini on [DMF] in this series, it is further necessary to know its net concentration in the sample solution, because the DMF molecules may be released from [UO2(TiPDGA)(DMF)2]2+ to give extra free DMF in the reaction system. To resolve this problem, the UV-vis absorption spectra of a CH2Cl2 solution dissolving [UO2(TiPDGA)(DMF)2]2+ were first recorded at different [Ac2O], where PhetOH was absent. As a result, little variation was observed as shown in Figure 4(a). It was confirmed that substitution of DMF with Ac2O does not proceed predominantly. Note that the result observed in this UV-vis titration experiment just describes thermodynamic stability of the preferred coordination of DMF to the U centre superior to that of Ac2O, and that it never rules out any possibility of the ligand substitution from DMF to Ac2O to form a reaction intermediate in the catalytic process of Figure 3.
We also collected UV-vis absorption spectra of the CH2Cl2 solution dissolving [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 (10 mM) at different [PhetOH], where Ac2O was absent. In contrast to the little dependency on [Ac2O] of Figure 4(a), significant spectral variation was observed with an increase in [PhetOH] as shown in Figure 4(b). Due to absence of any remarkable absorption of PhetOH at λ > 340 nm (Figure S5), substitution of the coordinated DMF by PhetOH has been confirmed. We have analyzed this spectral series with the HypSpec software [34] to assess the thermodynamic stabilities of the occurring species. Prior to this, the factor analysis [35] for the data set of Figure 4(b) revealed that this spectral variation consists of two principal components having meaningful eigenvalues (Figure S6), suggesting that two UO22+ complexes including the parent [UO2(TiPDGA)(DMF)2]2+ are present in the titration series observed in Figure 4(b). Consequently, the following ligand substitution equilibrium (Eq. (1)) with a logarithmic stability constant (log βsub) should be considered.
[UO2(TiPDGA)(DMF)2]2+ + PhetOH ⇄ [UO2(TiPDGA)(DMF)(PhetOH)]2+ + DMF log βsub
Although the second substitution of the remaining DMF molecule would also be possible, its contribution in Figure 4(b) should be negligibly small based on the above factor analysis of Figure S6. Eq. (1) is further divided into the following equilibria, Eqs. (2) and (3), to describe formation of [UO2(TiPDGA)(DMF)2]2+ and [UO2(TiPDGA)(DMF)(PhetOH)]2+, respectively.
[UO2(TiPDGA)]2+ + 2 DMF ⇄ [UO2(TiPDGA)(DMF)2]2+ log β20
[UO2(TiPDGA)]2+ + DMF + PhetOH ⇄ [UO2(TiPDGA)(DMF)(PhetOH)]2+ log β11
log βsub = log β11 – log β20
Herein, the desolvated UO22+ complex, [UO2(TiPDGA)]2+, is not an actual species occurring in the reaction system, but needs to be taken into account for expediency of the analysis.
As demonstrated by the 1H NMR spectroscopy (Figure S3), we have already confirmed that [UO2(TiPDGA)(DMF)2]2+ is present as a predominant species in the CD2Cl2 solution, indicating stability of this complex is high enough. Based on this fact, we derived log β11 of Eq. (3) from the spectral variation of Figure 4(b), where log β20 of Eq. (2) was fixed to an arbitrary value (e.g., log β20 = 10, 15, 20) large enough to guarantee the predominance of [UO2(TiPDGA)(DMF)2]2+ in the sample solution under absence of PhetOH. Consequently, the estimated log β11 was always smaller by 0.28 than log β20 with regardless of any above assumptions of log β20, i.e., log βsub of Eq. (1) is equal to −0.28. Such a preferential solvation of DMF compared with PhetOH should be ascribed to difference in the Lewis basicity between them pronounced by the donor numbers (DN) of DMF (26.6) and alcohols (~20) [31].
The calculated speciation diagram of [UO2(TiPDGA)(DMF)2]2+ and [UO2(TiPDGA)(DMF)(PhetOH)]2+ at different [PhetOH] (Figure S7) suggests that a mole fraction of [UO2(TiPDGA)(DMF)(PhetOH)]2+ is 0.85 at [PhetOH] = 0.1 M for instance. At the same time, one of DMF molecules initially present in [UO2(TiPDGA)(DMF)2]2+ is released in the same extent to give an extra concentration of DMF ([DMF]sub). Accordingly, the total concentration of free DMF ([DMF]total) in each sample solution of Figure 3(d) is sum of [DMF]add and [DMF]sub. When kini at different [DMF]add was plotted against [DMF]total−1, the linear relationship between these parameters was yielded (right panel of Figure 3(d)). Finally, kini is found to be first-order of [DMF]total−1 with a slope equal to 7.8 × 10−1 μM2⋅s−1.
In summary, the overall rate equation of the current catalytic acyl substitution studied in Figure 3 can be written down as follows.
where [U] denotes the concentration of the UO22+ catalyst, [UO2(TiPDGA)(DMF)2]2+ loaded. From the dependencies of kini with [Ac2O] and [DMF]total in Figure 3, the rate constant (k5) of Eq. (5) was estimated to be 1.6 × 10−4 s−1 at 27.5°C. Based on this rate equation and additional mechanistic insight obtained above, the catalytic cycle of the acyl substitution reaction of Ac2O with PhetOH mediated by [UO2(TiPDGA)(DMF)2]2+ is proposed as shown in Figure 5.
d[PhetOAc]/dt = kini = k5[Ac2O][DMF]−1[U]
As a preliminary step of this catalytic system, the ligand substitution reaction between [UO2(TiPDGA)(DMF)2]2+ and [UO2(TiPDGA)(DMF)(PhetOH)]2+ (Eq. (1)) is rapidly equilibrated in a reaction mixture, where the second ligand substitution to form [UO2(TiPDGA)(PhetOH)2]2+ is unlikely to occur probably due to steric hindrance between neighbouring PhetOH molecules. Ac2O enters the equatorial plane of UO22+ to replace one of the monodentate ligands. This is the rate-determining step of this catalytic reaction as defined by Eq. (5). It is hard to know which [UO2(TiPDGA)(DMF)2]2+ or [UO2(TiPDGA)(DMF)(PhetOH)]2+ is an actual activator of Ac2O, and which DMF or PhetOH is replaced by Ac2O for its activation. Because of these circumstances, the monodentate ligands of intermediate species in Figure 5 are simply denoted by “L”. Bidentate coordination of Ac2O instead of both Ls as observed in our former system of [UO2(OPPh3)4]2+ [22] is unlikely to occur, because kini is independent of [PhetOH] as shown in Figure 3(b) despite predominant formation of [UO2(TiPDGA)(DMF)(PhetOH)]2+ at the initial period of reaction as shown in Figure S7 based on log βsub of Eq. (1). The positively polarized carbonyl C of Ac2O after its coordination to the U centre readily undergoes nucleophilic attack from PhetOH to afford PhetOAc and AcOH. These reaction products are immediately removed from UO22+ due to weakly donating natures of esters (DN ~17) and carboxylic acids (DN = 10.5) [31]. Finally, the UO22+-TiPDGA complex returns to the initial state to repeat this catalytic cycle. The Lewis basicity of Ac2O is actually not very strong as pronounced by DN = 10.5 [31]. This should be one of the reasons why the reaction kinetics currently observed are commonly slow. In connection with this, the equatorial plane of UO22+ sterically constrained by tridentate TiPDGA would be another main cause of the slower reaction kinetics compared with that catalysed by [UO2(OPPh3)4]2+ we reported previously [22].
3. Conclusions
In this study, we have prepared a new UO22+ complex, [UO2(TiPDGA)(DMF)2]2+, bearing N,N,N’N’-tetraisopropyldiglycolamide (TiPDGA) and two DMF molecules to form a [3+1+1] coordination on the equatorial plane. The former auxiliary ligand provides a sterically regulated reaction field arising from its tridentate coordination manner, while the latter solvent molecules are reserved for ligand substitution with an acid anhydride to be activated after the coordination to the U centre at the initial stage of the reaction. Although several related UO22+-TRDGA complexes have been reported previously, the molecular structures of them are not very suitable for providing a Lewis-acidic reaction field required in this work. In contrast, the [3+1+1] equatorial coordination in the obtained [UO2(TiPDGA)(DMF)2]2+ was successfully attained by controlling preparation conditions such as stoichiometric loading of TiPDGA and employing the non-coordinating counteranion, ClO4−. Addition of extra HClO4 is also important to suppress undesired hydrolysis of UO22+. Catalytic activity of [UO2(TiPDGA)(DMF)2]2+ in the nucleophilic acyl substitution reactions of acid anhydrides was also examined. While the reaction rates are not very fast, some specific features such as suppression of the E1 side reaction of tBuOH and sterically constrained selectivity in activation of acid anhydrides have been observed. Based on the kinetic evaluation under different conditions, the reaction mechanism of this catalytic system has been proposed.
4. Experimental
4.1. Materials
Caution! All U isotopes are α-emitters. Therefore, standard precautions to handle radioactive materials should be followed. Perchlorate compounds are potentially explosive, so that such chemicals should be handled with great care. All chemicals except for U sources were of reagent grade and used as received, if not specified.
4.2. Synthesis of N,N,N’,N’-Tetraisoprolyldiglycolamide (TiPDGA)
Diglycolic dichloride (0.695 mL, 5.85 mmol, TCI) reacted with diisopropylamine (1.20 mL, 13 mmol, TCI) under presence of triethylamine (2.00 mL, 14 mmol, Wako) in CH2Cl2 (30 mL, Wako) at 0°C for 2 h, where the round-bottom flask reaction vessel was equipped with a CaCl2 drying tube. 1.2 M HCl(aq) (3 mL) was loaded to the reaction mixture, followed by stirring it for 5 min. After drying the separated organic layer over MgSO4, the filtrate was concentrated by a rotary evaporator. The residue was triturated with n-hexane (8 mL) under gentle heating to give colourless solid of TiPDGA (0.345 g, 20% yield). 1H NMR (CDCl3, δ/ppm vs. TMS): 4.22 (s, 4H OCH2), 3.92 (septet, 2H, CH(CH3)2), 3.45 (septet, 2H, CH(CH3)2), 1.40 (d, 12H, CH(CH3)2), 1.18 (d, 12H, CH(CH3)2). IR (ATR, diamond prism, cm−1) 1644 (νC=O).
4.3. Synthesis of [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2
UO3 (0.268 g, 0.936 mmol) from our in-house stock was dissolved in 70% HClO4 (180 μL, 2.1 mmol), followed by heating to concentrate it until white fume occurred. The cooled yellow residue of UO2(ClO4)2·xH2O was dissolved in triethyl orthoformate (10 mL). Under vigorous agitation at RT, DMF (305 μL, 3.96 mmol) was added dropwise to this mixture to afford bright yellow precipitate. The deposit was filtered out to obtain UO2(DMF)5(ClO4)2 (0.508 g, 0.609 mmol, 65% yield). This yellow compound further reacted with TiPDGA (0.294 g, 0.953 mmol) in CH2Cl2 (10 mL), where 70% HClO4 (10 μL) was also loaded to avoid hydrolysis of UO22+. After stirring for 2 h, this mixture was concentrated by slow evaporation in the dark. The yellow deposit was recrystallized from CH2Cl2 under Et2O vapor to give prismatic yellow crystals of [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2 (0.744 g, 80% yield).
When no 70% HClO4 was added to the CH2Cl2-based reaction mixture, the DMF-H2O mixed adduct, [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O, was crystallized together with yellow insoluble chunk precipitate instead of [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2.
Characterization of [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2
1H NMR (CD2Cl2, δ/ppm vs. TMS): 8.96 (s, 2H, CHO of DMF), 5.96 (s, 4H OCH2), 5.34 (s, 0.5H, CH2Cl2), 4.13 (septet, 2H, CH(CH3)2), 3.96 (septet, 2H, CH(CH3)2), 3.41-3.39 (s × 2, 12H, N(CH3)2 of DMF), 1.78 (d, 12H, CH(CH3)2), 1.46 (d, 12H, CH(CH3)2). IR (ATR, diamond prism, cm−1) 1644 (νC=O of DMF), 1607 (νC=O of TiPDGA), .934 (ν3 of UO22+). Anal. calcd for C23H48Cl4N4O15U: C 27.61, H 4.84, N 5.60; found: C 26.86, H 4.53, N 5.45. Crystallographic data for [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2. CCDC 2386158, Fw = 1000.48, 0.15 × 0.21 × 0.24 mm3, monoclinic, P21/n (#14), a = 11.1680(6) Å, b = 27.1647(11) Å, c = 13.5279(6) Å, β = 106.148(8)°, V = 3942.1(3) Å3, Z = 4, T = 183 K, Dcalcd = 1.686 g·cm−3, μ = 4.452 cm−1, GOF = 1.061, R (I > 2σ) = 0.0527, wR (all) = 0.0931.
Characterization of [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O
Crystallographic Data of [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O. CCDC 2386159, Fw = 878.49, 0.28 × 0.31 × 0.43 mm3, monoclinic, P21/n (#14), a = 13.5908(4) Å, b = 12.1091(3) Å, c = 20.5865(5) Å, β = 100.8350(10)°, V = 3327.57(15) Å3, Z = 4, T = 296 K, Dcalcd = 1.754 g·cm−3, μ = 5.106 cm−1, GOF = 1.099, R (I > 2σ) = 0.0289, wR (all) = 0.0703.
4.4. Methods
NMR spectra were recorded by a JEOL JNM ECX-400 spectrometer (1H: 399.78 MHz). IR spectra were measured by JSCO FT/IR-4700 equipped with a diamond ATR apparatus. CHN elemental analysis was performed using Yanaco CHN Corder MT-6. UV-vis absorption spectra were acquired by JASCO V-770 spectrophotometer. The structural characterization of UO22+ complexes has been carried out by single crystal X-ray diffraction. A single crystal of each sample was mounted on a Kapton capillary, and if necessary, located in the cryogenic N2 stream at the specified temperature. Intensity data were collected using Rigaku R-AXIS RAPID diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71075 Å). The obtained diffraction data were analyzed by Olex2 software package [36] suited with SHELX [37]. The structure was solved by SIR92, and expanded using Fourier techniques. All nonhydrogen atoms were anisotropically refined by SHELXL 2017/1 [38]. Hydrogen atoms were refined as riding on their parent atoms with Uiso(H) = 1.2Ueq(C). The final cycle of the full-matrix least-squares refinement of F2 was based on the observed reflections and parameters, and converged with the unweighted and weighted agreement factors, R and wR, respectively.
4.5. Catalytic Reaction
Nucleophilic acyl substitution of acid anhydrides catalyzed by [UO2(TiPDGA)(DMF)2]2+ was studied as follows. In an NMR tube, [UO2(TiPDGA)(DMF)2](ClO4)2·CH2Cl2 was dissolved in CD2Cl2 (700 μL), followed by loading a nucleophile and an acid anhydride appropriately. NMR spectra of each sample was recorded every 5-30 min to monitor progress of the reaction. During each experiment, temperature of the sample was maintained at 27.5°C with the temperature-controlled air flow in the NMR instrument.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figures S1 and S2: molecular structures of [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 and [UO2(TiPDGA)(DMF)(H2O)](ClO4)2⋅H2O; Figure S3: 1H NMR spectrum of [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 dissolved in CD2Cl2; Figure S4: molecular surface of [UO2(TiPDGA)(DMF)2]2+; Figure S5: UV-vis absorption spectra of CH2Cl2 solutions of PhetOH (100-600 mM); Figure S6: eigenvectors of principal components derived from the factor analysis for Figure 4(b); Figure S7: speciation diagram of UO22+-TiPDGA complexes (10 mM in total) at different [PhetOH] under presence of 20 mM DMF.
Author Contributions
Shin Akashi: Conceptualization, Data curation, Formal analysis, Investigation, Project administration, Writing – original draft, Writing – review & editing. Koichiro Takao: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
CCDC 2386158-2386159 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Takao, K. How does chemistry contribute to circular economy in nuclear energy systems to make them more sustainable and ecological? Dalton Trans. 2023, 52, 9866–9881. [Google Scholar] [CrossRef]
- Benedict, M.; Pigford, T.H.; Levi, H.W. Nuclear Chemical Engineering, 2nd ed.; McGraw-Hill: United States, 1981. [Google Scholar]
- Advanced Nuclear Fuel Cycles and Radioactive Waste Management; OECD-NEA: Paris, France, 2006.
- Actinide and Fission Product Partitioning and Transmutation, Status and Assessment Report; OECD-NEA: 1999.
- Actinide and Fission Product Partitioning and Transmutation, Eleventh Information Exchange Meeting; OECD-NEA: Issy-les-Moulineaux, France, 2012.
- Fujita, R.; Kawashima, M.; Ozawa, M.; Matsuzaki, T. Reduction and Resource Recycling of High-level Radioactive Wastes through Nuclear Transmutation -Overview and Current Progress-. JPS Conf. Proc. 2020, 32, 010098. [Google Scholar] [CrossRef]
- The Chemistry of the Actinide and Transactinide Elements; Morss, L. R., Edelstein, N.M., Fuger, J., Eds.; Springer: Netherlands, 2010. [Google Scholar]
- Barluzzi, L.; Giblin, S.R.; Mansikkamaki, A.; Layfield, R.A. Identification of Oxidation State +1 in a Molecular Uranium Complex. J. Am. Chem. Soc. 2022, 144, 18229–18233. [Google Scholar] [CrossRef]
- MacDonald, M.R.; Fieser, M.E.; Bates, J.E.; Ziller, J.W.; Furche, F.; Evans, W.J. Identification of the +2 oxidation state for uranium in a crystalline molecular complex, [K(2.2.2-cryptand)][(C5H4SiMe3)3U]. J. Am. Chem. Soc. 2013, 135, 13310–13313. [Google Scholar] [CrossRef]
- Billow, B.S.; Livesay, B.N.; Mokhtarzadeh, C.C.; McCracken, J.; Shores, M.P.; Boncella, J.M.; Odom, A.L. Synthesis and Characterization of a Neutral U(II) Arene Sandwich Complex. J. Am. Chem. Soc. 2018, 140, 17369–17373. [Google Scholar] [CrossRef] [PubMed]
- La Pierre, H.S.; Scheurer, A.; Heinemann, F.W.; Hieringer, W.; Meyer, K. Synthesis and characterization of a uranium(II) monoarene complex supported by delta backbonding. Angew. Chem. Int. Ed. 2014, 53, 7158–7162. [Google Scholar] [CrossRef] [PubMed]
- Barnea, E.; Eisen, M. Organoactinides in catalysis. Coord. Chem. Rev. 2006, 250, 855–899. [Google Scholar] [CrossRef]
- Liu, H.; Ghatak, T.; Eisen, M.S. Organoactinides in catalytic transformations: scope, mechanisms and Quo Vadis. Chem. Commun. 2017, 53, 11278–11297. [Google Scholar] [CrossRef]
- Cowie, B.E.; Purkis, J.M.; Austin, J.; Love, J.B.; Arnold, P.L. Thermal and Photochemical Reduction and Functionalization Chemistry of the Uranyl Dication, [UVIO2]2+. Chem. Rev. 2019, 119, 10595–10637. [Google Scholar] [CrossRef] [PubMed]
- Behera, N.; Sethi, S. Unprecedented Catalytic Behavior of Uranyl(VI) Compounds in Chemical Reactions. Eur. J. Inorg. Chem. 2020, 2021, 95–111. [Google Scholar] [CrossRef]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Takao, K.; Takao, S.; Ikeda, Y.; Bernhard, G.; Hennig, C. Uranyl-halide complexation in N,N-dimethylformamide: halide coordination trend manifests hardness of [UO2]2+. Dalton Trans. 2013, 42, 13101–13111. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, S.; Liu, K.; Yuan, L.; Zhao, Y.; Chai, Z.; Mei, L. Facile construction of diverse diarylmethane scaffolds via uranyl-catalyzed 1,6-addition reaction. Tetrahedron Lett. 2020, 61, 152076. [Google Scholar] [CrossRef]
- van Axel Castelli, V.; Cort, A.D.; Mandolini, L.; Reinhoudt, D.N. Supramolecular Catalysis of 1,4-Thiol Addition by Salophen−Uranyl Complexes. J. Am. Chem. Soc. 1998, 120, 12688–12689. [Google Scholar] [CrossRef]
- van Axel Castelli, V.; Dalla Cort, A.; Mandolini, L.; Reinhoudt, D.N.; Schiaffino, L. Catalysis fo the Addition of Benzenethiol to 2-Cyclohexene-1-ones by Uranyl-Salophen Complexes: A Catalytic Metalloclreft with High Substrate Specificity. Chem. Eur. J. 2000, 6, 1193–1198. [Google Scholar] [CrossRef]
- Dalla Cort, A.; Mandolini, L.; Schiaffino, L. Exclusive transition state stabilization in the supramolecular catalysis of Diels-Alder reaction by a uranyl salophen complex. Chem. Commun. 2005, 3867–3869. [Google Scholar] [CrossRef]
- Takao, K.; Akashi, S. Exploring the catalytic activity of Lewis-acidic uranyl complexes in the nucleophilic acyl substitution of acid anhydrides. RSC Adv. 2017, 7, 12201–12207. [Google Scholar] [CrossRef]
- Monsigny, L.; Thuéry, P.; Berthet, J.-C.; Cantat, T. Breaking C–O Bonds with Uranium: Uranyl Complexes as Selective Catalysts in the Hydrosilylation of Aldehydes. ACS Catal. 2019, 9, 9025–9033. [Google Scholar] [CrossRef]
- Yu, J.; Chen, S.; Liu, K.; Yuan, L.; Mei, L.; Chai, Z.; Shi, W. Uranyl-catalyzed hydrosilylation of para-quinone methides: access to diarylmethane derivatives. Org. Biomol. Chem. 2021, 19, 1575–1579. [Google Scholar] [CrossRef]
- Sasaki, Y.; Choppin, G.R. Solvent Extraction of Eu, Th, U, Np and Am with N,N’-Dimethyl-N,N’-dihexyl-3-oxapentanediamide and Its Analogous Compounds. Anal. Sci. 1996, 12, 225–230. [Google Scholar] [CrossRef]
- Kannan, S.; Moody, M.A.; Barnes, C.L.; Duval, P.B. Lanthanum(III) and uranyl(VI) diglycolamide complexes: synthetic precursors and structural studies involving nitrate complexation. Inorg. Chem. 2008, 47, 4691–4695. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, K.; Kurahashi, K.; Naganawa, H. Extraction behavior of lanthanides using a diglycolamide derivative TODGA in ionic liquids. Dalton Trans. 2008, 5083–5088. [Google Scholar] [CrossRef]
- Kawasaki, T.; Okumura, S.; Sasaki, Y.; Ikeda, Y. Crystal Structures of Ln(III) (Ln = La, Pr, Nd, Sm, Eu, and Gd) Complexes with N,N,N′,N′-Tetraethyldiglycolamide Associated with Homoleptic [Ln(NO3)6)]3−. Bull. Chem. Soc. Jpn. 2014, 87, 294–300. [Google Scholar] [CrossRef]
- Tian, G.; Rao, L.; Teat, S.J.; Liu, G. Quest for environmentally benign ligands for actinide separations: thermodynamic, spectroscopic, and structural characterization of U(VI) complexes with Oxa-diamide and related ligands. Chem. Eur. J. 2009, 15, 4172–4181. [Google Scholar] [CrossRef]
- Ansari, S.A.; Wadawale, A.P.; Verboom, W.; Mohapatra, P.K. Isolation of single crystals of a homoleptic UO22+-diglycolamide complex from a room temperature ionic liquid: X-ray crystallography and complexation studies. New J. Chem. 2022, 46, 950–954. [Google Scholar] [CrossRef]
- Linert, W.; Fukuda, Y.; Camard, A. Chromotropism of coordination compounds and its applications in solution. Coord. Chem. Rev. 2001, 218, 113–152. [Google Scholar] [CrossRef]
- Deshayes, L.; Keller, N.; Lance, M.; Nierlich, M.; Vigner, D. Structure of pentakis(N,N-dimethylformamide)dioxouranium(VI) tetrafluoroborate. Acta Crystallogr. 1992, C48, 1660–1661. [Google Scholar] [CrossRef]
- Takao, K.; Ikeda, Y. Structural characterization and reactivity of UO2(salophen)L and [UO2(salophen)]2: dimerization of UO2(salophen) fragments in noncoordinating solvents (salophen = N,N'-disalicylidene-o-phenylenediaminate, L = N,N-dimethylformamide, dimethyl sulfoxide). Inorg. Chem. 2007, 46, 1550–1562. [Google Scholar] [CrossRef]
- Sabatini, A.; Vacca, A.; Gans, P. Mathematical algorithms and computer programs for the determination of equilibrium constants from potentiometric and spectrophotometric measurements. Coord. Chem. Rev. 1992, 120, 389–405. [Google Scholar] [CrossRef]
- Rossberg, A.; Reich, T.; Bernhard, G. Complexation of uranium(VI) with protocatechuic acid-application of iterative transformation factor analysis to EXAFS spectroscopy. Anal. Bioanal. Chem. 2003, 376, 631–638. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
Figure 1.
Lewis-acidic UO22+ catalysts employed in the former works (L: solvent molecule) (a) [18,19,20,21,22,23,24] and our strategy for molecular design of UO22+ catalyst having [3+1+1] equatorial plane sterically regulated by an auxiliary tridentate TRDGA ligand together with two L molecules reserved for substrate activation (b).
Figure 1.
Lewis-acidic UO22+ catalysts employed in the former works (L: solvent molecule) (a) [18,19,20,21,22,23,24] and our strategy for molecular design of UO22+ catalyst having [3+1+1] equatorial plane sterically regulated by an auxiliary tridentate TRDGA ligand together with two L molecules reserved for substrate activation (b).
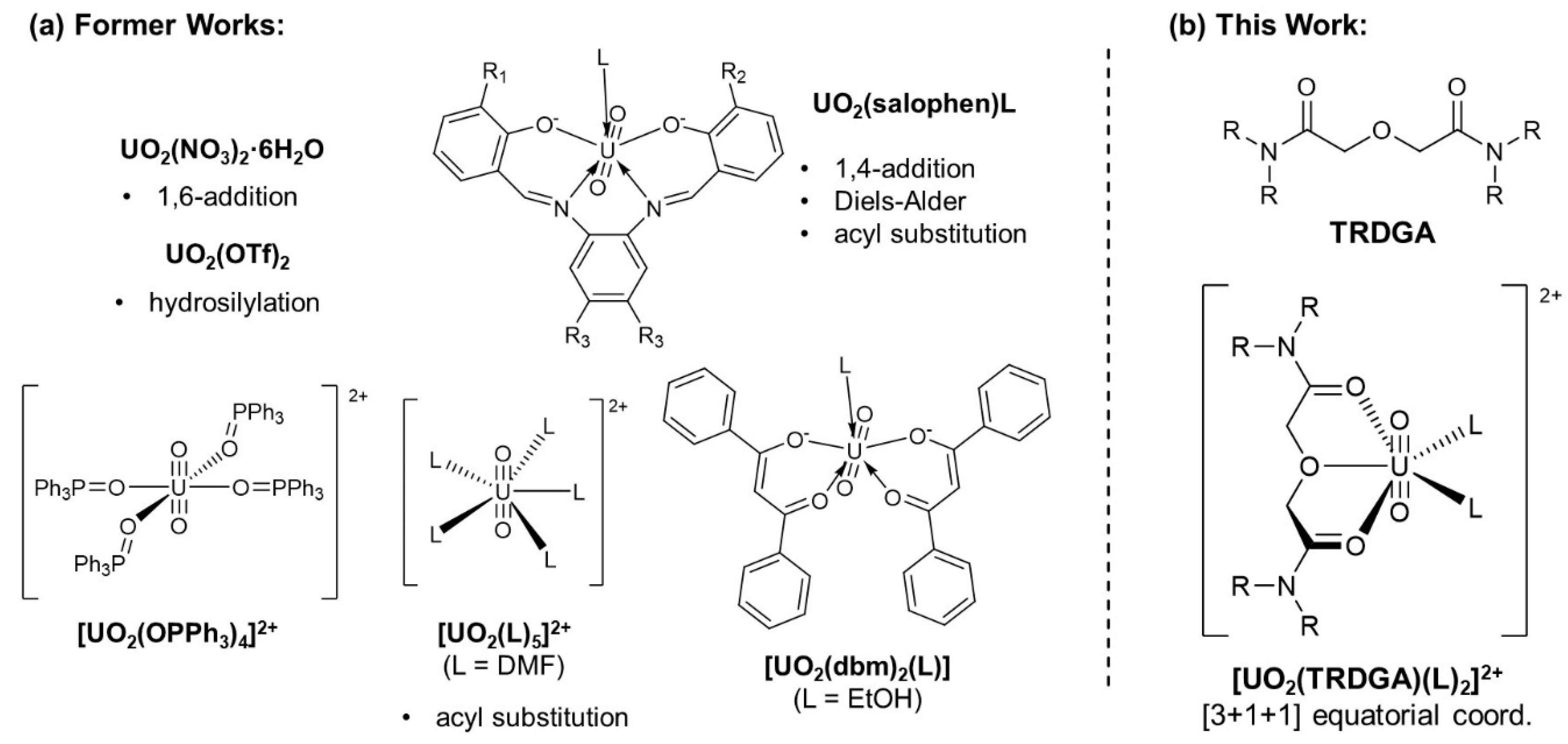
Figure 2.
Molecular structure of [UO2(TiPDGA)(DMF)2]2+ in its perchlorate salt with a CH2Cl2 molecule as a crystalline solvent. Thermal ellipsoids are drawn in 50% probability level. Hydrogen atoms were omitted for clarity. Bond distances (Å): U(1)−O(1) 1.751(4), U(1)−O(2) 1.750(4), U(1)−O(3) 2.520(3), U−O(4) 2.354(4), U−O(5) 2.344(3), U(1)−O(6) 2.325(4), U(1)−O(7) 2.343(4). Bond angles (°): O(1)−U(1)−O(2) 179.46(19), O(3)−U(1)−O(4) 61.51(11), O(3)−U(1)−O(5) 61.74(11), O(5)−U(1)−O(6) 77.07(13), O(4)−U(1)−O(7) 80.50(13), O(6)−U(1)−O(7) 79.46(14), Σeq = 360.28.
Figure 2.
Molecular structure of [UO2(TiPDGA)(DMF)2]2+ in its perchlorate salt with a CH2Cl2 molecule as a crystalline solvent. Thermal ellipsoids are drawn in 50% probability level. Hydrogen atoms were omitted for clarity. Bond distances (Å): U(1)−O(1) 1.751(4), U(1)−O(2) 1.750(4), U(1)−O(3) 2.520(3), U−O(4) 2.354(4), U−O(5) 2.344(3), U(1)−O(6) 2.325(4), U(1)−O(7) 2.343(4). Bond angles (°): O(1)−U(1)−O(2) 179.46(19), O(3)−U(1)−O(4) 61.51(11), O(3)−U(1)−O(5) 61.74(11), O(5)−U(1)−O(6) 77.07(13), O(4)−U(1)−O(7) 80.50(13), O(6)−U(1)−O(7) 79.46(14), Σeq = 360.28.
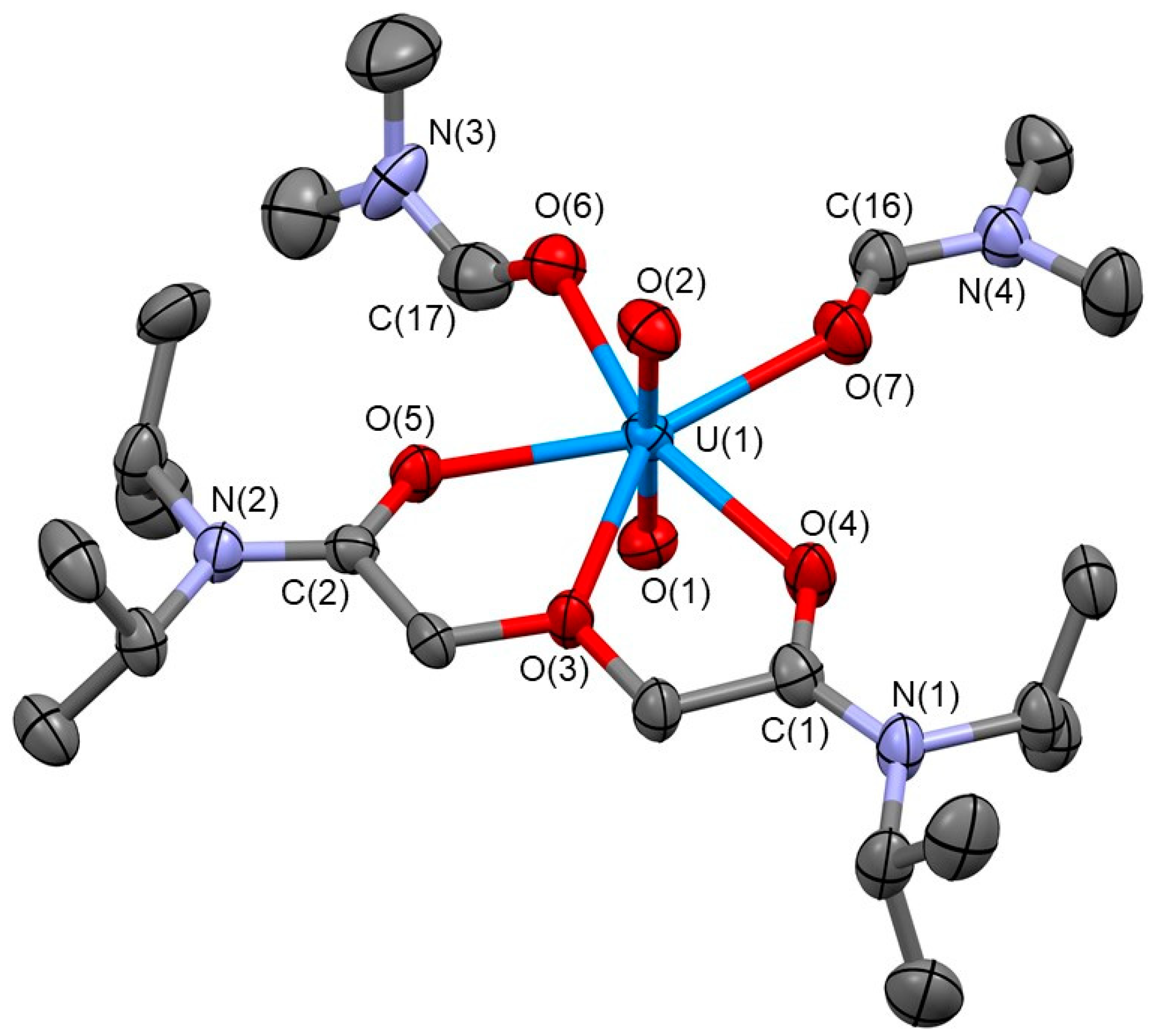
Figure 3.
Kinetic study on nucleophilic acyl substitution of acetic anhydride (Ac2O) with 2-phenylethanol (PhetOH) to afford 2-phenylethyl acetate (PhetOAc) and acetic acid (AcOH) under presence of [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 (10 mM, 2 mol%) in CD2Cl2 at 27.5°C. (a) Typical traces of reactants (Ac2O, PhetOH) and product (PhetOAc) with elapse of time, where 506 mM Ac2O and 480 mM PhetOH were loaded at the initial state. (b) Dependency on initial [PhetOH]. (c) Dependency on initial [Ac2O] together with initial rate (kini) as a function of [Ac2O]. (d) Dependency on concentration of DMF additionally loaded to the reaction system ([DMF]add) together with kini as a function of reciprocal concentration of total DMF present in the sample solution ([DMF]total−1). Based on ligand substitution reaction (Eq. (2)) with log βsub, a mole fraction of [UO2(TiPDGA)(DMF)(PhetOH)]2+ is 0.96 at [PhetOH] = 0.50 M (see Figure S7), where DMF is also released to the solution in the same extent to give extra DMF concentration ([DMF]sub). Accordingly, [DMF]total = [DMF]add + [DMF]sub.
Figure 3.
Kinetic study on nucleophilic acyl substitution of acetic anhydride (Ac2O) with 2-phenylethanol (PhetOH) to afford 2-phenylethyl acetate (PhetOAc) and acetic acid (AcOH) under presence of [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 (10 mM, 2 mol%) in CD2Cl2 at 27.5°C. (a) Typical traces of reactants (Ac2O, PhetOH) and product (PhetOAc) with elapse of time, where 506 mM Ac2O and 480 mM PhetOH were loaded at the initial state. (b) Dependency on initial [PhetOH]. (c) Dependency on initial [Ac2O] together with initial rate (kini) as a function of [Ac2O]. (d) Dependency on concentration of DMF additionally loaded to the reaction system ([DMF]add) together with kini as a function of reciprocal concentration of total DMF present in the sample solution ([DMF]total−1). Based on ligand substitution reaction (Eq. (2)) with log βsub, a mole fraction of [UO2(TiPDGA)(DMF)(PhetOH)]2+ is 0.96 at [PhetOH] = 0.50 M (see Figure S7), where DMF is also released to the solution in the same extent to give extra DMF concentration ([DMF]sub). Accordingly, [DMF]total = [DMF]add + [DMF]sub.
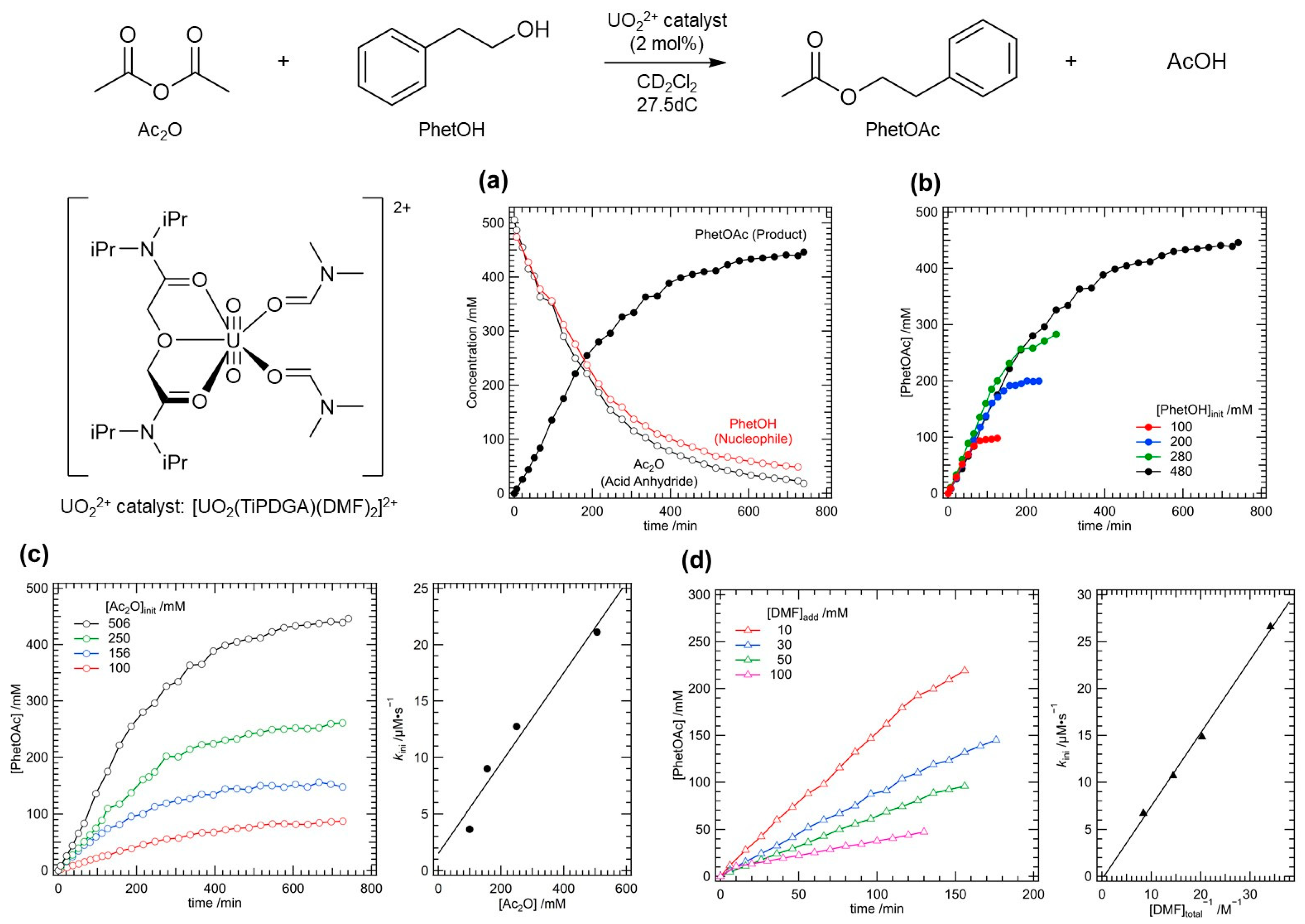
Figure 4.
UV-vis absorption spectra of CH2Cl2 solutions dissolving [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 (10 mM) acquired at different [Ac2O] (a) and [PhetOH] (b).
Figure 4.
UV-vis absorption spectra of CH2Cl2 solutions dissolving [UO2(TiPDGA)(DMF)2](ClO4)2⋅CH2Cl2 (10 mM) acquired at different [Ac2O] (a) and [PhetOH] (b).
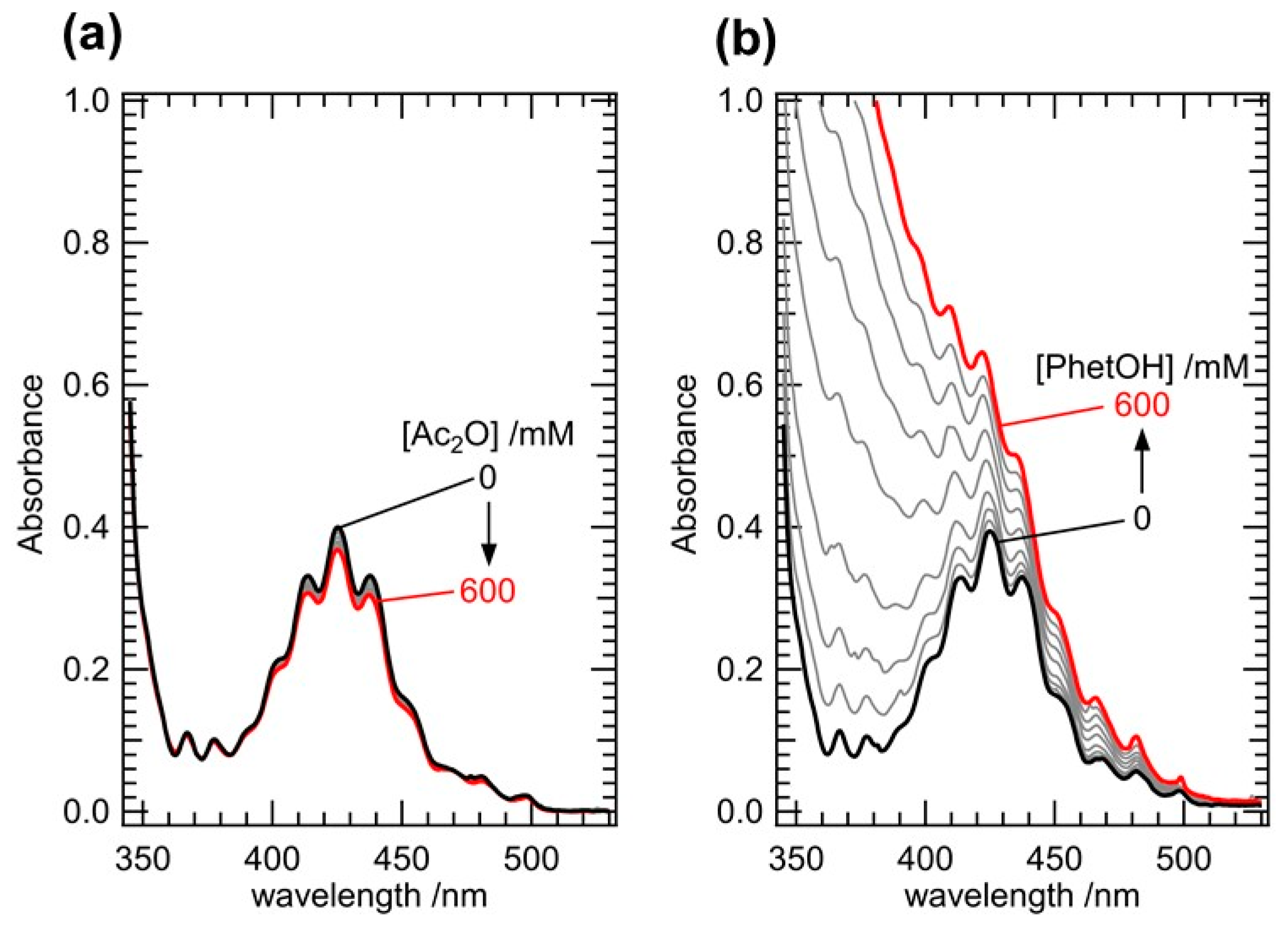
Figure 5.
Proposed reaction mechanism of the nucleophilic acyl substitution reaction of Ac2O catalysed by [UO2(TiPDGA)(DMF)2]2+, where a nucleophile is PhetOH (R’ = 2-phenylethyl). The electric charges on the UO22+ complexes are omitted for clarity. L: monodentate ligand such as DMF or PhetOH.
Figure 5.
Proposed reaction mechanism of the nucleophilic acyl substitution reaction of Ac2O catalysed by [UO2(TiPDGA)(DMF)2]2+, where a nucleophile is PhetOH (R’ = 2-phenylethyl). The electric charges on the UO22+ complexes are omitted for clarity. L: monodentate ligand such as DMF or PhetOH.
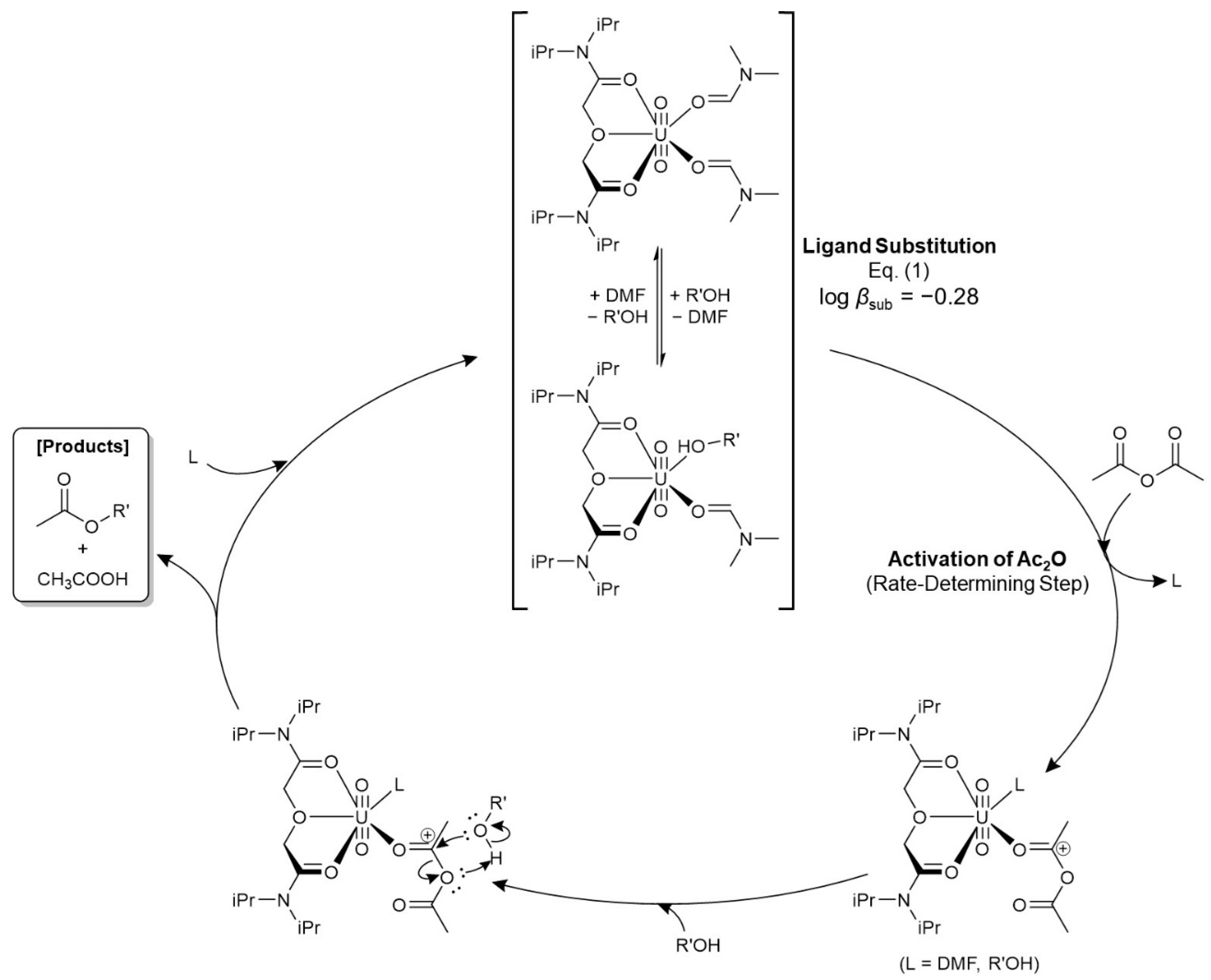

Table 1.
Summary of nucleophilic acyl substitution reactions of acid anhydrides ((RCO)2O + R’OH → RCOOR’ + RCOOH) catalysed by [UO2(TiPDGA)(DMF)2]2+ in CD2Cl2a.
Table 1.
Summary of nucleophilic acyl substitution reactions of acid anhydrides ((RCO)2O + R’OH → RCOOR’ + RCOOH) catalysed by [UO2(TiPDGA)(DMF)2]2+ in CD2Cl2a.
| entry | R b | R’ c | yield (%)d |
| 1 | Me | 2-phenylethyl | 90 |
| 2 | Me | 2-phenylethyl | 9e |
| 3 | Me | Me | 100 |
| 4 | Me | Et | 88 |
| 5 | Me | nBu | 88 |
| 6 | Me | Ph | 95 |
| 7 | Me | tBu | 78 |
| 8 | Et | 2-phenylethyl | 85 |
| 9 | iPr | 2-phenylethyl | 48 |
| 10 | tBu | 2-phenylethyl | 10 |
| 11 | Ph | 2-phenylethyl | 6 |
| 12 | tBu | Me | 14 |
a Typical condition: 0.5 M acid anhydride + 0.5 M nucleophile + 10 mM catalyst (2.0 mol%), if not specified. b R of acid anhydrides. c R’ of nucleophiles. d Determined by 1H NMR peak integrals. e No catalyst loaded.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



