1. Introduction
Hepatoblastoma and medulloblastoma are two rare, aggressive pediatric cancers of embryonic origin that present unique challenges for treatment due to their underlying genetic complexity and shared developmental pathways [
1,
2]. These tumors frequently involve dysregulation in the Wnt/β-catenin signaling pathway, which is essential for embryonic development and the maintenance of stem cell populations [
3,
4,
5]. Mutations in CTNNB1, which encodes β-catenin, are commonly observed in both hepatoblastoma and certain medulloblastoma subtypes, particularly those classified as WNT-subtypes, highlighting a shared oncogenic mechanism that implicates Wnt signaling in the progression and characteristics of these cancers [
6,
7].
Central to Wnt pathway activity is the transcription factor LEF1, part of the TCF/LEF family, which serves as a key mediator of β-catenin signaling [
8,
9]. LEF1 drives the expression of target genes associated with cellular proliferation, differentiation, and survival, functions that are often hijacked in cancer to promote tumor growth [
10,
11]. Recent findings suggests that LEF1’s influence extends beyond traditional Wnt pathway signaling, impacting metabolic and epigenetic landscapes within tumors [
12]. These roles, however, remain incompletely understood, particularly in the context of a shared LEF1-mediated transcriptional program across hepatoblastoma and medulloblastoma.
This study aims to bridge this gap by investigating the common transcriptional and molecular features driven by LEF1 in both hepatoblastoma and WNT-subtype medulloblastoma. Through integrative bioinformatics and multi-omics analyses at both the tumor and single-cell levels, we identify a robust WNT-LEF1 gene expression signature enriched in tumor cells from both cancers. This signature, comprising critical regulatory genes, highlights a conserved embryonic transcriptional pro-gram likely contributing to tumor progression and therapeutic resistance. By elucidating the LEF1-dependent mechanisms within the Wnt pathway, our findings provide new insights into the molecular underpinnings of these two pediatric tumors and reveal potential targets for therapeutic intervention that leverage this shared oncogenic axis.
2. Materials and Methods
2.1. Public Transcriptome Datasets
2.1.1. MB-PBTA (MB, OpenPBTA)
The Open Pediatric Brain Tumor Atlas [
13]: RNA-sequencing data for medulloblastoma from the PBTA were downloaded from the Pediatric cBioPortal website, part of the “Open Pediatric Cancer Project” consortium [
14], at the address:
https://pedcbioportal.kidsfirstdrc.org/ (accessed on November 6, 2024). After filtration and clinical annotation, this transcriptomic medulloblastoma cohort consisted of 254 tumor samples, with most samples from male patients (61%) and primary tumors (61%). These tumors were distributed among distinct molecular subtypes: WNT (n = 21), SHH (n = 74), G3 (n = 60), and G4 (n = 99) (
Table 1). RNA-sequencing counts were transformed into pseudocounts using a log2 + 1 transformation for downstream analyses.
2.1.2. GSE37418 (MB)
Normalized data matrices and annotations (GPL570-[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) from dataset GSE37418 were downloaded from the Gene Expression Omnibus (GEO) website [
15] using the GEOquery R package version 2.70.0 [
16] in the R software environment version 4.3.3. This transcriptomic cohort of medulloblastoma tumors included 74 tumor samples, with the majority from male patients (71%) and a mean age of 99 months. Most patients were of white ethnicity (57%), followed by Hispanic ethnicity (18%). Regarding molecular subtypes, this cohort consisted of 39 G4 samples, 8 WNT samples, 11 SHH samples, and 16 G3 samples (
Table 2).
2.1.3. GSE44971 (Normal Cerebellum)
Normalized data matrices and annotations (GPL570-[HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array) from dataset GSE44971 [
17] were downloaded from the GEO website [
15] using the GEOquery R package version 2.70.0 [
16] in the R software environment version 4.3.3. This cohort comprised samples of normal cerebellar tissue.
2.1.4. GSE104766 (HB and Normal Liver)
RNA-sequencing normalized counts (Illumina HiSeq 2500, Homo sapiens) were downloaded from the GEO NCBI website at
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE104766 (accessed on November 6, 2024). This cohort included 30 hepatoblastoma tumor samples and 30 normal liver samples. No associated clinical data were provided for this cohort.
2.1.5. GSE131329 (HB)
This RNA-seq transcriptomic cohort comprised 14 samples of noncancerous liver tissue and 53 samples of hepatoblastoma tumors. Of the pathological samples, 30 were histologically well-differentiated, and 20 were poorly differentiated. The mean age of the patients at the time of sampling was 27 months. Each PRETEXT stage (1–4) was well-balanced among the study samples, and the majority of samples exhibited beta-catenin genomic alterations (
Table 3).
2.2. Public Single Cell Transcriptome Datasets
2.2.1. scRNA-seq Medulloblastoma
The GSE155446 dataset comprised single-cell RNA-sequencing data from 28 pediatric medulloblastoma samples, stratified into the four medulloblastoma subtypes: SHH, WNT, G3, and G4. For these experiments, libraries were prepared using Chromium Single Cell V2 and V3 Chemistry Library Kits (10× Genomics). Barcoded cDNA was sequenced on an Illumina NovaSeq 6000, achieving 50,000 reads per cell. Data were demultiplexed using CellRanger (10× Genomics) [
18]. Raw counts were integrated into a single-cell object using Seurat version 5.1.0. The standard Seurat pipeline, including NormalizeData, FindVariableFeatures, ScaleData, and RunPCA, was applied to the single-cell object [
19]. A single-cell expression score was computed using a thirteen-gene signature with the AddModuleScore function in Seurat.
2.2.2. scRNA-seq Hepatoblastoma:
Single-cell RNA-sequencing data from the GEO dataset GSE180665 [
20] were downloaded from the following web address:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE180665 (accessed on August 1, 2024). This dataset included seven scRNA-seq experiments performed on three hepatoblastoma tumors, two patient-derived xenotransplantation (PDX) models, and two adjacent liver tissue samples. Raw counts in H5AD format were converted into a single-cell experiment object using the zellkonverter R-Bioconductor package [
21]. The original cell annotation was added to the metadata of the scRNA-seq object after its conversion into a Seurat object [
19].
2.3. ChIP-Sequencing in Human Pluripotent H1 Cell Line
To characterize LEF1 chromatin binding occupancy in humans at the embryonic stage within the context of the bivalent promoter program [
22], different ChIP-sequencing datasets performed on H1 human pluripotent stem cells were accessed from the Gene Expression Omnibus (GEO) database:
GSM1579343: LEF1 untreated; Homo sapiens with SRA alias SRR1745491,
GSM1579344: LEF1 Wnt3a; Homo sapiens with SRA alias SRR1745492,
GSM1693959: Input H1; Homo sapiens with SRA alias SRR2037029,
GSM1579348: H3K27ac Wnt3a; Homo sapiens with SRA alias SRR1745496,
GSM1579350: H3K27me3 Wnt3a; Homo sapiens with SRA alias SRR1745498.
These experiments were conducted by the Jones Lab in San Diego, California [
23]. For ChIP-seq, DNA was extracted using the QIAquick PCR extraction kit (cat 28106, Qiagen). Single-end reads were sequenced on an Illumina HiSeq 2500. The ChIP-sequencing data were downloaded in FASTQ format from the NCBI database using the fastq-dump command from the SRA Toolkit (version 3.1.1) [
24].
2.4. Transcriptome Cross Normalization for Common Signature Between Hepatoblastoma and Medulloblastoma Tumors
Transcriptome analyses were conducted in the R software environment (version 4.3.3). To investigate common regulatory mechanisms in hepatoblastoma and medulloblastoma, a cross-normalized matrix was constructed by merging distinct transcriptome datasets: GSE37418, comprising medulloblastoma tumor samples; GSE44971, comprising normal cerebellum controls; and GSE104766, comprising hepatoblastoma tumors and normal liver samples.
To account for experimental batch effects, cross-normalization was performed using the Combat function from the SVA R package (version 3.50.0) [
25]. Following the Combat processing step, quantile normalization was applied to reduce noise between samples using the PreprocessCore R package (version 1.64.0) [
26].
For downstream analyses, the LIMMA (Linear Models for Microarray Data) R package (version 3.58.1) was employed to identify differentially expressed genes between tumor samples (hepatoblastoma and medulloblastoma) and their respective controls (normal liver and normal cerebellum) [
27]. Functional enrichment analyses of significant genes were conducted using the clusterProfiler R package (version 4.10.1) [
28,
29] with Gene Ontology Molecular Function and Biological Process databases [
30].
2.5. ChIP-Sequencing Analyses
Chromatin immunoprecipitation sequencing (ChIP-seq) experiments were analyzed following the recommended guidelines of the ENCODE consortium [
31]. Downloaded FASTQ files were assessed for quality using FastQC (version 0.74). FASTQ files were trimmed with Trimmomatic (version 0.39) [
32], and the quality of the trimmed reads was subsequently evaluated with FastQC.
Reads were aligned to the HG38 human genome using Bowtie2 (version 2.5.3) in very sensitive end-to-end mode [
33]. BAM files were sorted using SAMtools (version 1.19.2) [
34] and filtered for mapping quality scores greater than 30 with BamTools (version 2.5.2) [
35]. Narrow peaks representing LEF1 genome binding in H1 cells stimulated with WNT3A were called using the MACS2 peak caller (version 2.2.9.1) [
36,
37], with H1 input chromatin serving as the background control [
38].
MACS2-generated narrow peaks were annotated with HOMER (version 4.11) [
39] on the HG38 genome, using the GencodeV46 annotation file [
40]. Promoter-annotated regions from the resulting BED file were filtered for downstream analysis. BigWig files were generated using the bamCoverage command from deepTools (version 3.5.4) [
41]. Genomic heatmaps of promoter regions were created using the double commands computeMatrix and plotHeatmap from deepTools.
Functional enrichment analyses of significant genes were conducted using the clusterProfiler R package (version 4.10.1) [
28,
29] with Gene Ontology Molecular Function and Biological Process databases [
30].
2.6. WNT-LEF1 Expression Score
Using the expression of 13 WNT-LEF1 target genes in the medulloblastoma PBTA cohort, univariate logistic regression analyses were conducted using the logitloop R package (version 1.0.0), available at
https://github.com/cdesterke/logitloop (accessed on November 13, 2024). The binomial status of the WNT subtype was used as the outcome variable for the regression analyses.
A WNT-LEF1 expression score was subsequently calculated by summing the expression levels of the target genes weighted by the binomial beta coefficients obtained from the univariate analyses. The optimal cutpoint for the expression score was determined using the cutpointr R package (version 1.1.2) [
42].
2.7. ElasticNet Modeling on WNT-LEF1 Expression Signature
The expression levels of the 13 WNT-LEF1 target genes were extracted from medulloblastoma tumors and combined with the corresponding WNT subtype status as metadata. After splitting the data into training and validation sets in a 0.6/0.4 ratio, an ElasticNet model (with tumor cell status as the binary outcome) was tuned for alpha and lambda parameters using the caret R package (version 6.0-94) [
42]. The final ElasticNet model was fitted with the optimal alpha parameter (alpha = 0.1) using the glmnet R package (version 4.1-8) [
43].
3. Results
3.1. LEF1 Upregulation as a Shared Molecular Feature in Hepatoblastoma and Medulloblastoma
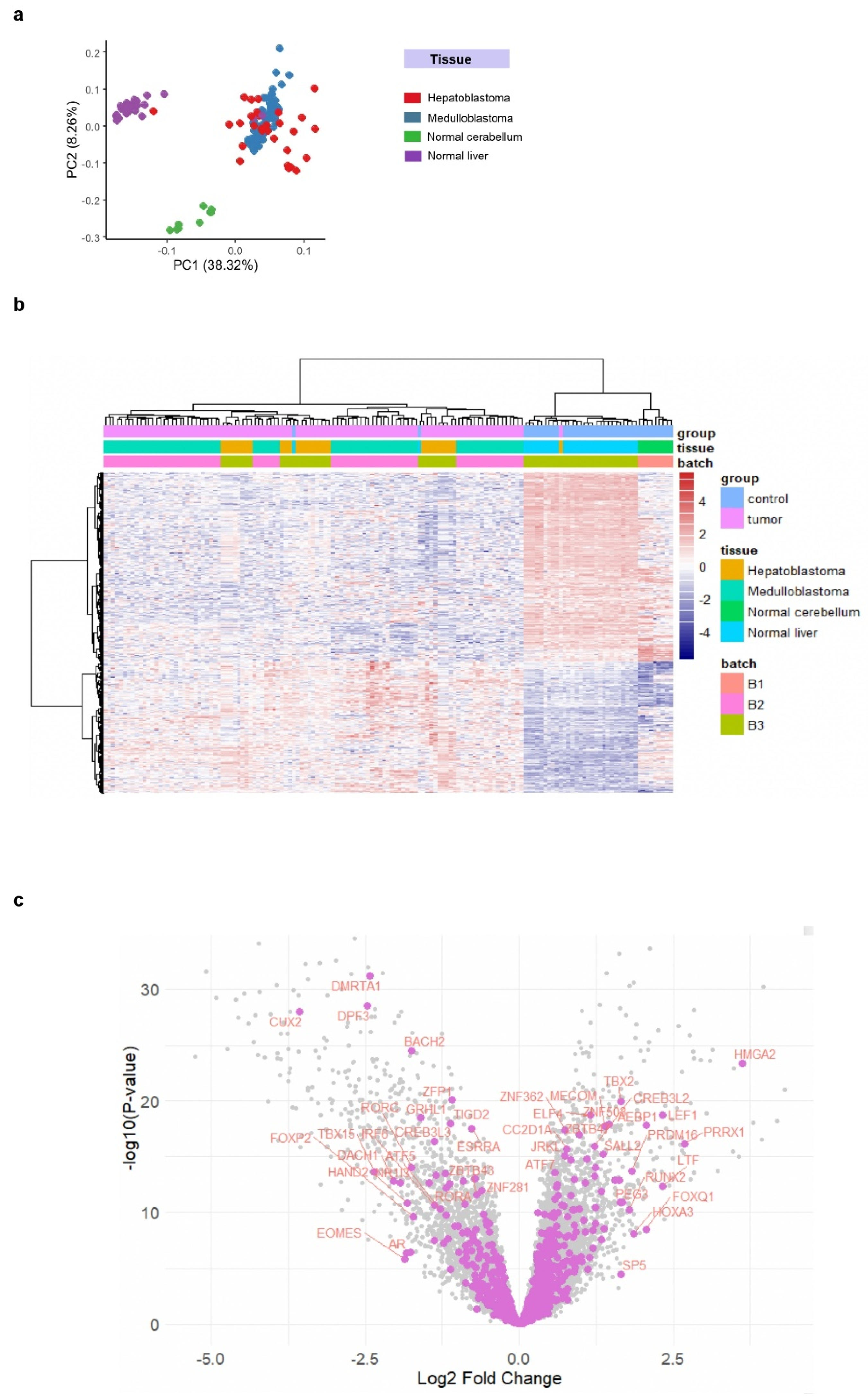
Hepatoblastoma and medulloblastoma are two types of pediatric tumors with embryonic origins. Tumor cells in these two types frequently exhibit genetic alterations in WNT pathway genes, particularly involving mutations in CTNNB1. Based on these shared characteristics, we constructed a combined transcriptome dataset that includes medulloblastoma and hepatoblastoma tumor samples, along with control samples from the respective tissues of origin (cerebellum for medulloblastoma and adjacent healthy liver for hepatoblastoma).
To conduct this analysis, we assembled data from the GSE37418 (medulloblastoma), GSE44971 (cerebellum control), and GSE104766 (normal liver and hepatoblastoma) datasets, normalizing them in a common matrix using the Combat algorithm to adjust for batch effects (
Supplemental Figure S1A). The heterogeneity in Combat density plots across samples (
Supplemental Figure S1B) was reduced by applying quantile post-Combat normalization (
Supplemental Figure S1C). Unsupervised principal component analysis (PCA) of the fully normalized transcriptome data confirmed that samples from different batches were well integrated (
Supplemental Figure S2A). Additionally, this analysis clearly distinguished tumor samples (medulloblastoma and hepatoblastoma) from normal controls (cerebellum and liver) (
Supplemental Figure S2B), suggesting a shared transcriptional expression signature between these tumors.
To identify this common transcriptional signature, we performed a supervised differential expression analysis between tumor and control samples using the LIMMA algorithm. With a significance threshold of |log2 fold change| ≥ 1 and an FDR-adjusted p-value < 0.05, we identified 1259 differentially expressed genes (DEGs) significantly regulated in both hepatoblastoma and medulloblastoma compared to their respective controls (Volcano plot in
Supplemental Figure S2C; list of DEGs in
Supplemental Table S1). Unsupervised PCA of these 1259 DEGs successfully stratified tumor samples from controls on the first principal component (
Figure 1A). This tumor-specific clustering was further confirmed by unsupervised hierarchical clustering using Euclidean distances and the ward.D2 method (
Figure 1B).
Functional enrichment analysis using the Gene Ontology Molecular Function (GO-MF) database revealed that the 513 upregulated genes in tumors were predominantly associated with extracellular matrix interactions, including structural components (e.g., collagens), integrin binding, collagen binding, and growth factor binding (
Supplemental Figure S3A,B). Conversely, analysis of the 746 downregulated genes in tumors highlighted a significant enrichment in vitamin-interacting molecules and enzymes with aromatase activity, such as cytochrome P450 family members (
Supplemental Figure S4A,B).
Using Toronto’s database of human transcription factors [
44], we projected transcription factor identifiers onto the volcano plot (
Figure 1C) to examine their regulation in tumors. Notable upregulated transcription factors included HMGA2, TBX2, CREB3L2, and LEF1 (
Figure 1C and
Supplemental Table S2). Among these, ChIP-sequencing data on LEF1 binding in human embryonic H1 stem cells have been previously conducted [
45]. This epigenetic information allows us to investigate the LEF1-dependent transcriptional program that operates during human embryonic development.
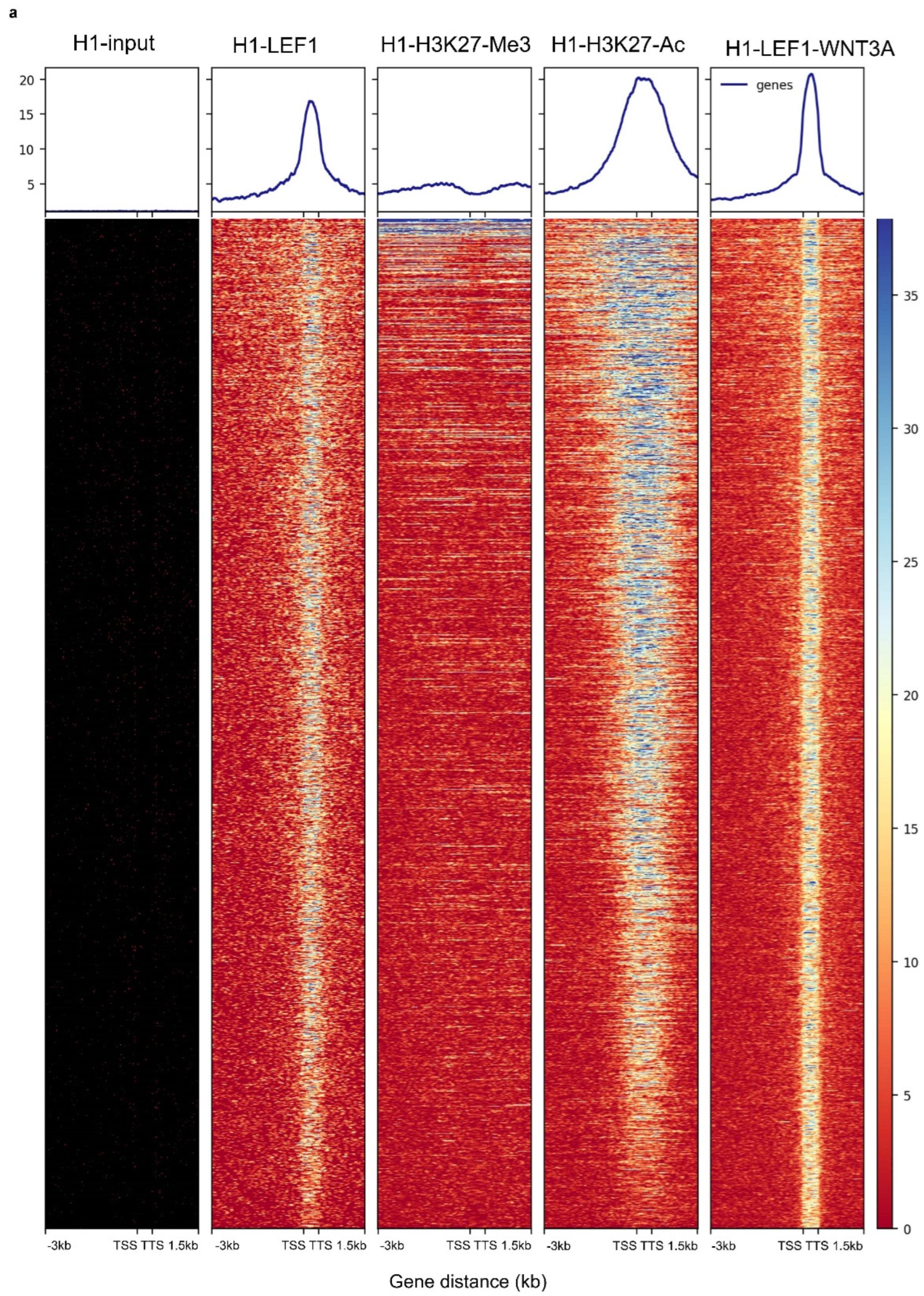
3.2. WNT3A Stimulation Activates LEF1 Chromatin Binding Program with H3K27ac Co-Occupancy in H1 Embryonic Cells
LEF1 ChIP-sequencing experiment conducted on H1 pluripotent human stem cells after WNT3A stimulation was aligned to the HG38 human genome using the Bowtie2 algorithm. Similar ChIP-sequencing experiments were also performed for H1 input chromatin and LEF1 in H1 cells without WNT3A stimulation. Additionally, the bivalency of pluripotent promoter regions was examined using ChIP-sequencing for histone marks H3K27me3 (a repressive mark) and H3K27ac (an active mark), following the same pipeline. Narrow peak calling for regions with high-quality mapped reads (Q > 30) was carried out using the MACSII algorithm, with H1 input chromatin as a control for peak calling in LEF1-H1-WNT3A ChIP-seq. This analysis identified 11,436 enriched genomic regions.
After annotating peaks with Homer, 2039 were found to be in proximal promoter regions (
Supplemental Table S3). For these 2039 promoter regions, a genomic matrix was generated using Deeptools, centered around Transcription Start Sites (TSS) on the HG38 genome, extending 3 kb upstream and 1.5 kb downstream. This analysis included five ChIP-sequencing experiments: H1 input, H1-LEF1, H1 H3K27me3, H1 H3K27ac, and H1-LEF1-WNT3A. The resulting genomic heatmap (Figure 3) confirmed the presence of these promoter regions in the H1-LEF1-WNT3A ChIP-seq experiment, with no corresponding peaks in the H1 input chromatin ChIP-seq. These enrichments were also observed in the H1-LEF1 ChIP-seq experiment, though with reduced intensity compared to the WNT3A stimulation condition. Notably, these promoter regions largely coincided with active histone marks, specifically H3K27ac (
Figure 2). Through this analysis, an active LEF1 embryonic chromatin binding program was identified in H1 stem cells upon stimulation with the WNT pathway ligand WNT3A.
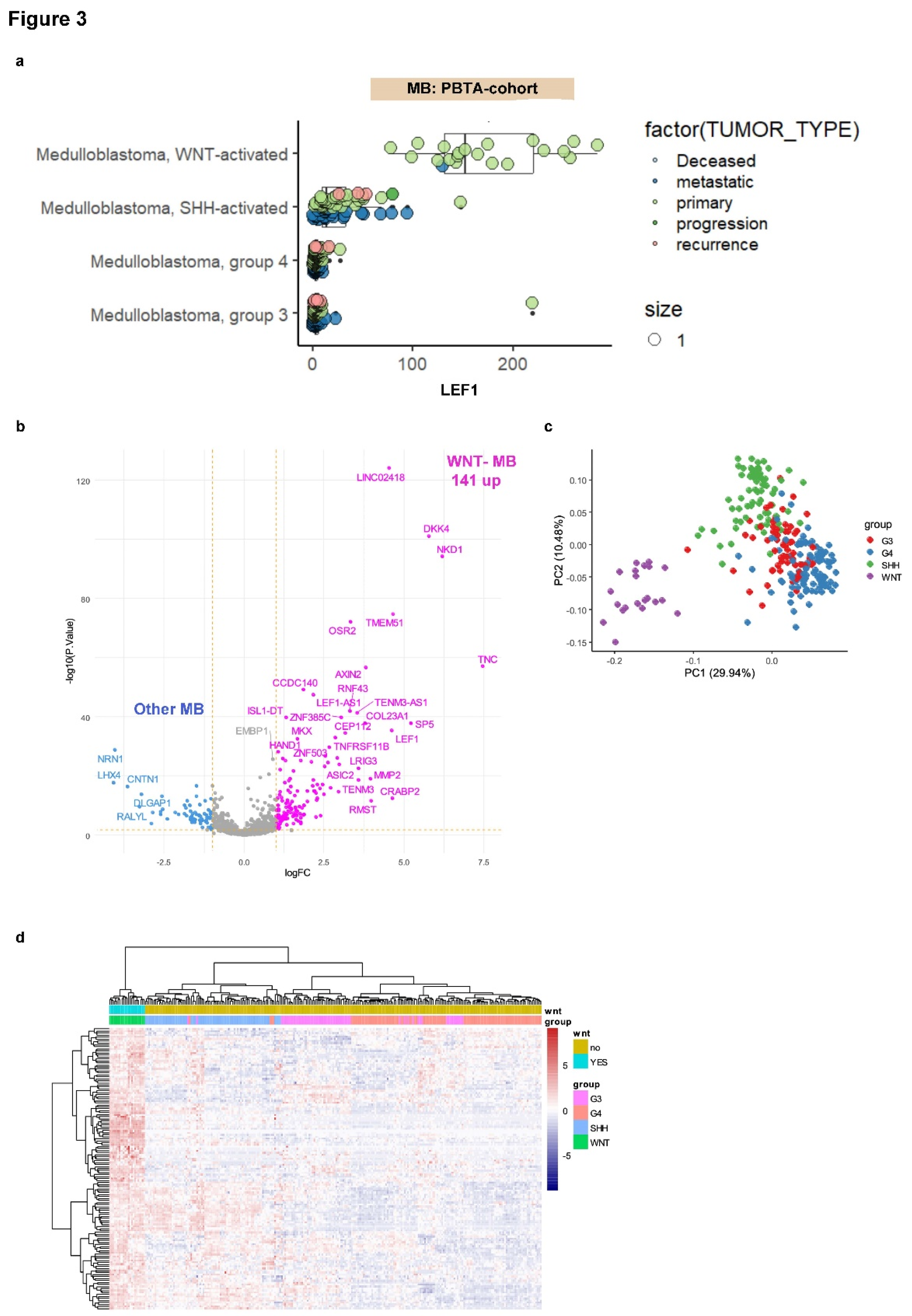
3.3. LEF1 Embryonic Transcriptional Network Is a Hallmark of WNT-Subtype Medulloblastoma
In embryonic medulloblastoma, heterogeneity in WNT pathway activation has been characterized through transcriptome analysis [
23,
46]. The Pediatric Brain Tumor Atlas (PBTA) cohort [
47] was filtered to include RNA sequencing data from medulloblastoma samples classified by molecular subtypes: WNT, SHH, Group 3 (G3), and Group 4 (G4) (n = 254;
Table 1). LEF1 expression was analyzed according to the molecular subtype classification within the PBTA cohort (
Figure 3A). LEF1 was found to be significantly increased in the transcriptomes of WNT subtype medulloblastomas compared to other subtypes (ANOVA p ≤ 2E-16), predominantly in samples categorized as primary tumors (
Figure 3A).
LEF1 target genes, identified through ChIP-sequencing as 2039 promoter regions (
Supplemental Table S3), were mapped onto the transcriptome data of medulloblastoma samples in the PBTA cohort. A supervised differential expression analysis was performed between WNT subtype tumors and other molecular subtypes to assess the regulation of LEF1 target genes. Using a significance threshold of |log2 fold change| ≥ 1 and FDR-adjusted p-value < 0.05, 207 DEGs were identified, with the majority (n = 141) being overexpressed in WNT subtype tumors compared to other medulloblastoma subtypes (
Figure 3B;
Supplemental Table S4). Principal component analysis (PCA) based on the expression of these 141 LEF1 target genes effectively stratified WNT subtype tumors from other medulloblastoma subtypes (
Figure 3C). This stratification was further validated using unsupervised hierarchical clustering with Euclidean distances and the Ward.D2 method (
Figure 3D).
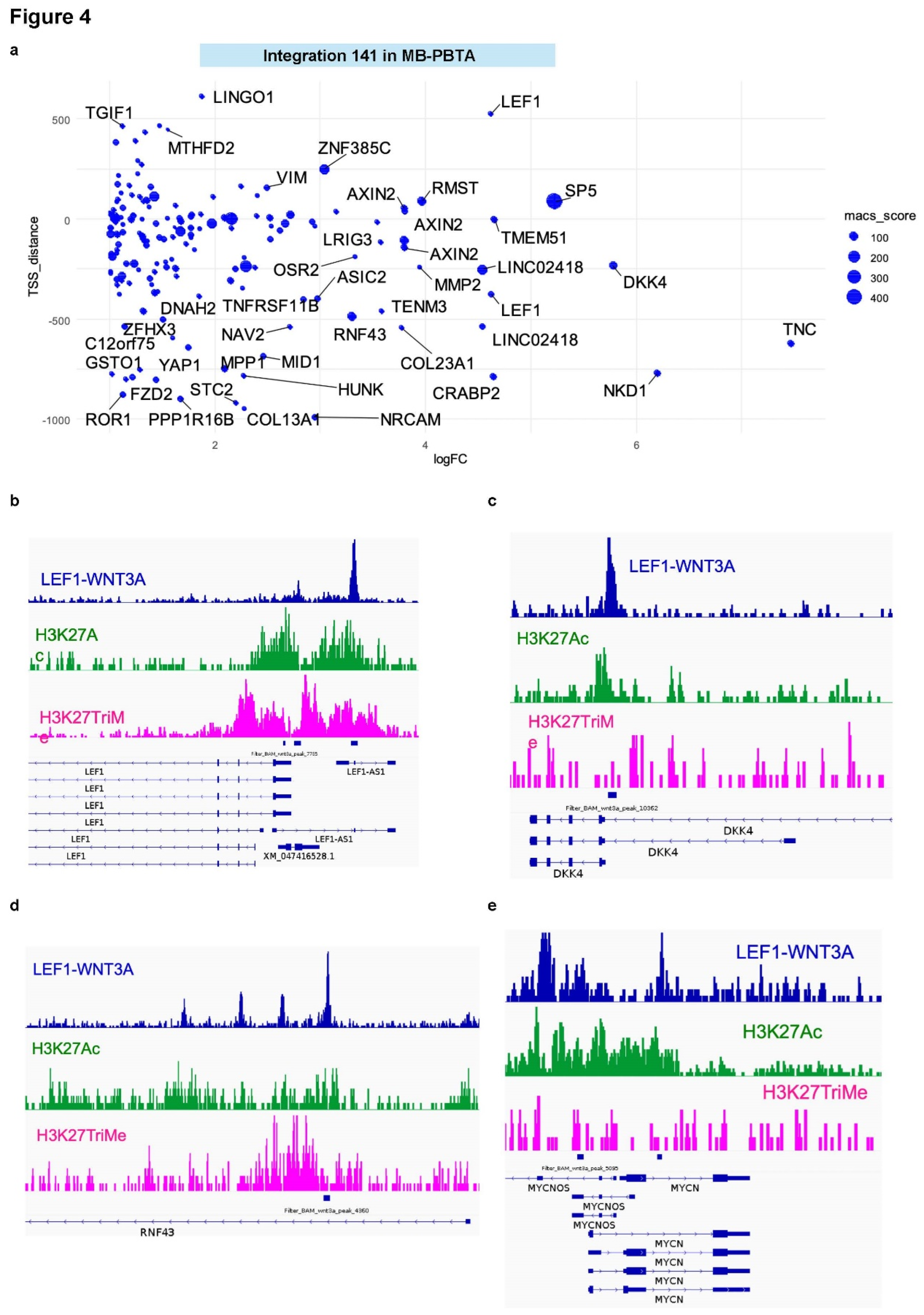
Functional enrichment analysis (Gene Ontology Biological Process [GO-BP] database) of the 141 LEF1 target genes highlighted their roles in mesenchymal development, embryonic organ development, and WNT pathway regulation (Supplemental Figure S5A; Figure 4A). Notably, bivalent histone marks (positive for both H3K27ac and H3K27me3) were detected at the genomic loci of LEF1/LEF1-AS1 (
Figure 4B) and RNF43 (a WNT pathway component and LEF1 target;
Figure 4D). Active histone marks (H3K27ac) were also found at the promoters of DKK4 (WNT pathway regulator and LEF1 target;
Figure 4C) and MYCN (a LEF1 target;
Figure 3E).
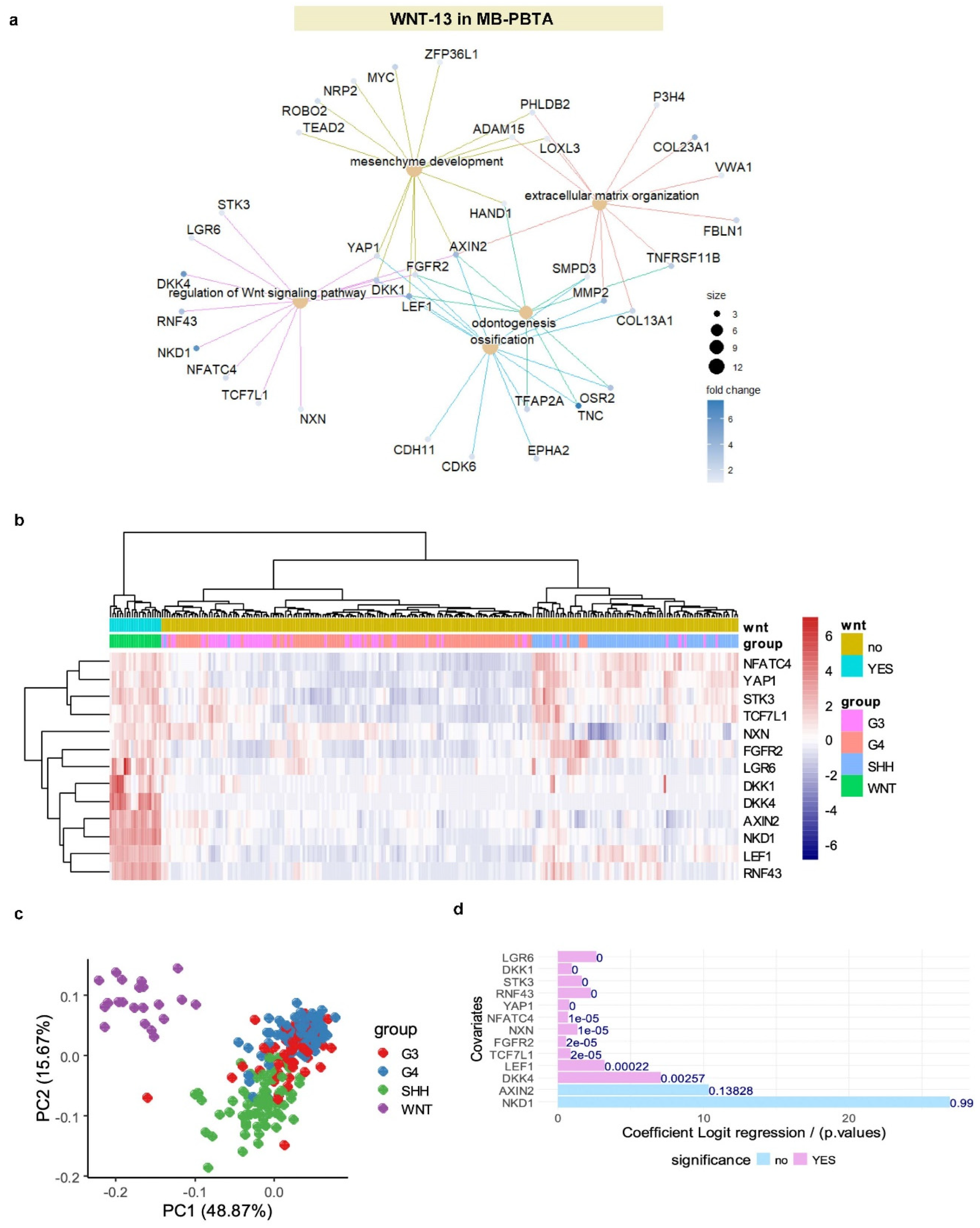
Among the LEF1 targets upregulated in WNT-medulloblastoma, 13 genes were identified as regulators of the WNT pathway (
Figure 5A). These included RNF43 (ring finger protein 43), LEF1 (lymphoid enhancer binding factor 1), NKD1 (NKD inhibitor of WNT signaling pathway 1), AXIN2 (axin 2), DKK4 (dickkopf WNT signaling pathway inhibitor 4), DKK1 (dickkopf WNT signaling pathway inhibitor 1), LGR6 (leucine-rich repeat-containing G protein-coupled receptor 6), FGFR2 (fibroblast growth factor receptor 2), NXN (nucleoredoxin), TCF7L1 (transcription factor 7 like 1), STK3 (serine/threonine kinase 3), YAP1 (Yes1 associated transcriptional regulator), and NFATC4 (nuclear factor of activated T cells 4). Using the expression profiles of these 13 WNT-related LEF1 target genes, unsupervised clustering (Euclidean distances and Ward.D2 method) effectively stratified WNT-medulloblastoma samples from some samples in other medulloblastoma subtypes in the PBTA cohort (
Figure 5B). This stratification was corroborated by PCA (
Figure 5C), and univariate binomial analysis with WNT-medulloblastoma status as the outcome confirmed significant associations for 11 of the 13 genes (
Figure 5D).
ROC curve analysis of the combined expression of these 13 WNT-related LEF1 targets yielded an area under the curve (AUC) of 1.00, indicating 100% specificity and sensitivity for predicting the WNT subtype in the PBTA medulloblastoma cohort (
Supplemental Figure S5B). Based on the expression and binomial beta-coefficients of the 13 WNT-LEF1 genes (
Figure 5D), a WNT-LEF1 expression score was computed for medulloblastoma samples in the PBTA cohort. This analysis identified 21 samples with high WNT-LEF1 expression scores, compared to 233 remaining samples with lower scores (
Table 1). Between the high- and low-score groups, no significant gender bias was observed (p = 1.00). However, high-score samples were primarily categorized as primary tumors (p = 0.023), predominantly belonged to the WNT subtype (p = 1E-4) and showed an increased proportion of tumors sampled from the ventricle area (p = 0.039) (
Table 1).
3.4. WNT-LEF1 Gene Signature Accurately Predicts WNT Subtype in Medulloblastoma Transcriptomes
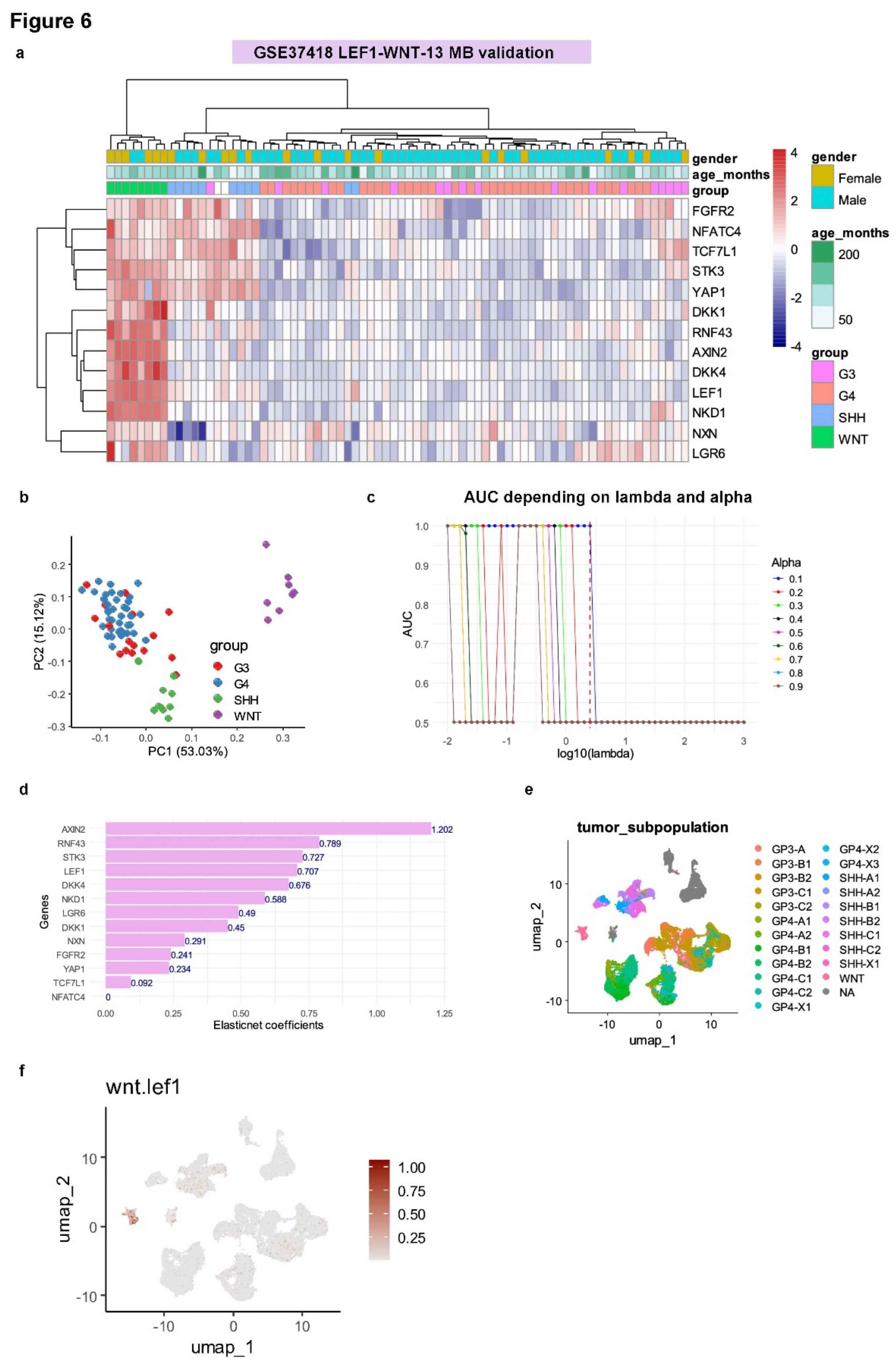
For validation of the thirteen-gene signature, an independent medulloblastoma transcriptome cohort (GSE37418;
Table 2) was analyzed. This cohort included 74 tumor samples, with most patients being male (72%) and a mean age at sampling of 99 months. Unsupervised clustering based on the expression of the thirteen WNT-LEF1 genes effectively stratified WNT samples from other medulloblastoma subtypes (Euclidean distances and Ward.D2 method;
Figure 6A). This stratification was further confirmed using unsupervised principal component analysis (
Figure 6B).
Supervised machine learning tuned using ElasticNet regression (alpha = 0.1;
Figure 6C and
Supplemental Figure S6A,B) allowed us to predict WNT-subtype status with a perfect area under the curve (AUC) of 1.00 (
Figure 6C). ElasticNet coefficients (
Figure 6D) highlighted the predictive importance of genes such as AXIN1, RNF43, STK3, LEF1, and DKK1 for identifying WNT subtype status in this medulloblastoma validation cohort.
A WNT-LEF1 expression score was computed for this validation cohort, identifying 8 samples with high WNT-LEF1 expression scores and 66 samples with low scores (
Table 2). In this cohort, patients with high WNT-LEF1 expression scores were significantly more likely to be female (p = 0.007) and perfectly matched the WNT subtype (
Table 2 and
Supplemental Figure S6C). ROC curve analysis of the combined expression of these thirteen WNT-related LEF1 target genes confirmed their ability to predict WNT subtype in medulloblastoma, achieving an AUC of 1.00 with 100% specificity and 100% sensitivity in the PBTA medulloblastoma cohort (
Supplemental Figure S6D).
Finally, to investigate the single-cell heterogeneity of the signature in medulloblastoma cells, scRNA-seq experiments from 28 medulloblastoma tumors representing the four distinct subtypes (GSE155446;
Figure 6E) were analyzed for the expression of the 13 WNT-LEF1 genes. The single-cell WNT-LEF1 expression score was found to be positive exclusively in the cluster of WNT tumor cells (
Figure 6F). At the single-cell level in medulloblastoma, the WNT-LEF1 thirteen-gene signature was expressed only in tumor cells from the WNT subtype.
3.5. Single-Cell Analysis Validates WNT-LEF1 Signature Activation in Hepatoblastoma Tumor Cells
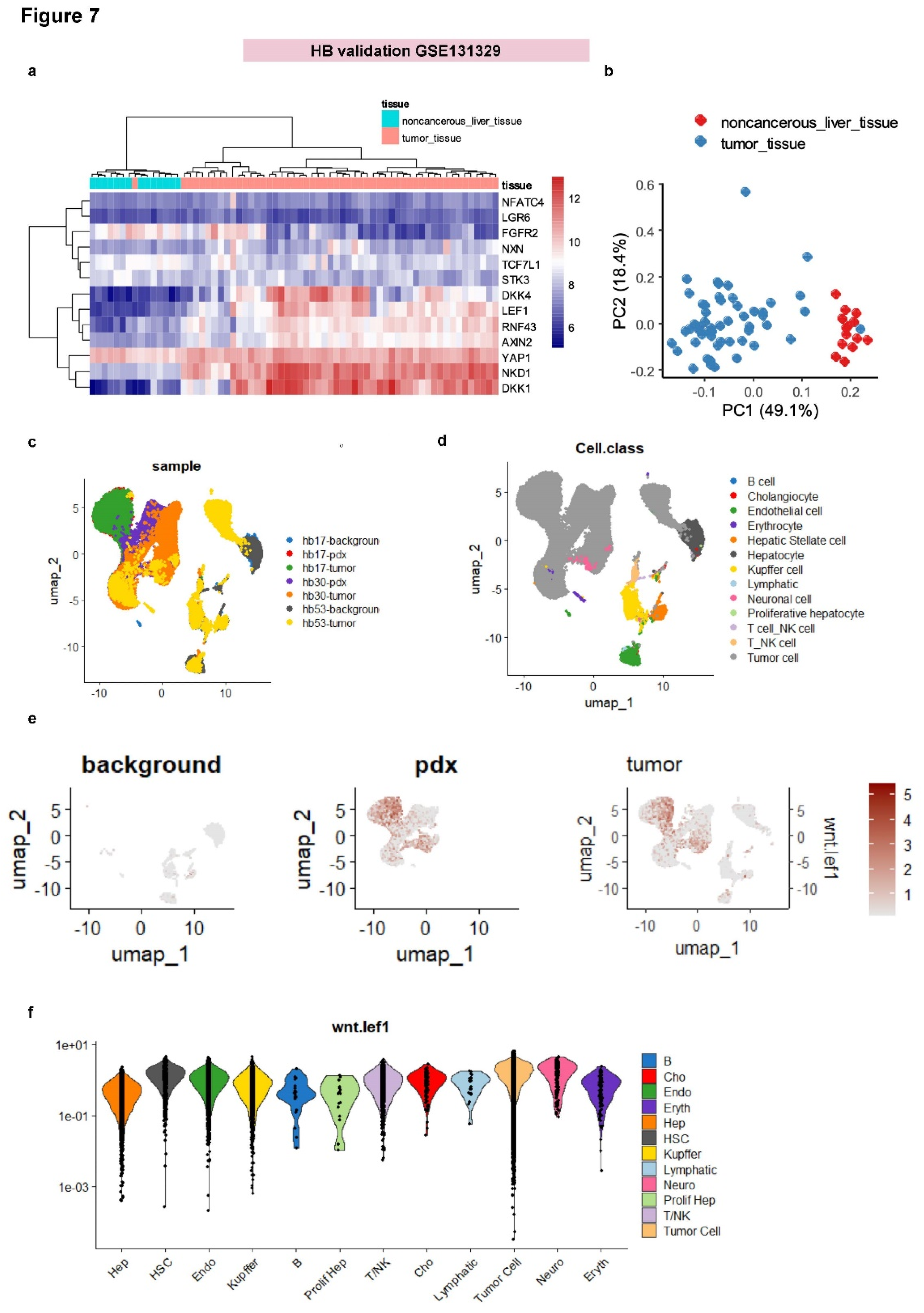
Like medulloblastoma, hepatoblastoma is also of embryonic origin, and tumor cells are frequently affected by genomic alterations in the CTNNB1 gene [
48]. The expression of the 13 genes from the WNT-LEF1 signature was analyzed in the hepatoblastoma transcriptome cohort GSE131329 (
Table 3), which includes 14 noncancerous liver samples and 53 hepatoblastoma tumors. Unsupervised clustering based on the expression of the 13-gene signature effectively stratified hepatoblastoma tumor samples from noncancerous liver tissue (Euclidean distances and Ward.D2 method;
Figure 7A). This stratification was further validated by unsupervised principal component analysis (
Figure 7B).
The WNT-LEF1 signature showed potential for stratifying clinical parameters of hepatoblastoma, such as histological differentiation and PRETEXT stages (data not shown). Notably, more than 70% of tumors in this cohort exhibited genetic alterations in the CTNNB1 gene (
Table 3). To identify the specific cell types in which the WNT-LEF1 signature was activated, single-cell transcriptome data from human and PDX hepatoblastoma samples, compared to normal adjacent liver tissue, were analyzed (GSE180655;
Figure 7C). Thirteen distinct cell types were characterized in these samples (
Figure 7D).
The WNT-LEF1 single-cell expression score was computed for cells in these experiments. The score was negative in normal adjacent liver tissue but positive in PDX and tumor samples (
Figure 7E). This score was confirmed to be significantly higher in tumor cells (
Figure 7F and
Table 4). At the single-cell level, the expression of the WNT-LEF1 13-gene signature was confirmed to be activated in tumor cells from human hepatoblastoma.
4. Discussion
During this study, the transcription factor LEF1 was found to be commonly upregulated in hepatoblastoma and medulloblastoma tumors. While LEF1 deregulation in hepatoblastoma remains poorly understood, in the PBTA medulloblastoma cohort, high LEF1 expression was specifically associated with the WNT subtype. Patients with WNT-subtype medulloblastoma generally exhibit better prognoses than those with other subtypes [
49]. LEF1 immunohistochemistry has been recognized as a more reliable diagnostic biomarker than β-catenin for identifying WNT-activated medulloblastoma subtypes [
50,
51]. Additionally, LEF1, together with TCF3, has been implicated in maintaining the self-renewal of mouse embryonic stem cells [
52].
An integrative analysis of WNT-LEF1 targets in the PBTA medulloblastoma cohort identified 141 LEF1-activated genes specific to the WNT subtype. Functional enrichment revealed that 13 of these genes were involved in WNT pathway regulation, while others were mainly associated with mesenchymal development and extracellular matrix organization. LEF1 is known to facilitate epithelial-mesenchymal transition (EMT), a hallmark of cancer progression characterized by increased migration and invasion of tumor cells [
53,
54,
55].
Among LEF1 targets, MYCN was identified as a direct target through promoter binding. During embryonic development, the MYCN promoter exhibits the H3K27-acetylation histone mark, indicating active chromatin at this stage. MYCN is a proto-oncogene encoding a basic helix-loop-helix transcription factor that is upregulated in hepatoblastoma and other pediatric liver tumors. It plays a critical role in promoting hepatoblastoma cell proliferation [
56]. In medulloblastoma, genomic amplification of MYCN is routinely detected during diagnosis [
57].
Wnt signaling is a key developmental pathway crucial for embryonic development and adult stem cell maintenance, and its dysregulation is implicated in numerous diseases. Through multi-omics analyses, we identified a WNT-LEF1 integrative signature that accurately predicts the WNT subtype of both medulloblastoma and hepatoblastoma tumors. This signature includes RNF43 (ring finger protein 43), LEF1 (lymphoid enhancer binding factor 1), NKD1 (NKD inhibitor of WNT signaling pathway 1), AXIN2 (axin 2), DKK4 (dickkopf WNT signaling pathway inhibitor 4), DKK1 (dickkopf WNT signaling pathway inhibitor 1), LGR6 (leucine-rich repeat-containing G protein-coupled receptor 6), FGFR2 (fibroblast growth factor receptor 2), NXN (nucleoredoxin), TCF7L1 (transcription factor 7 like 1), STK3 (serine/threonine kinase 3), YAP1 (Yes1 associated transcriptional regulator), and NFATC4 (nuclear factor of activated T cells 4).
RNF43 is a transmembrane E3 ligase that removes Wnt receptors from the stem cell surface and acts as a negative feedback regulator of the WNT pathway [
58]. AXIN2 and NKD1 are also negative feedback regulators in Wnt signaling, with AXIN2 destabilizing cytoplasmic β-catenin and NKD1 inhibiting its nuclear localization [
59]. Among the five Wnt antagonists identified, DKK4 binds to lipoprotein receptor-related protein 5/6 (LRP5/6) and Kremen, inducing LRP endocytosis to block β-catenin signal transduction [
60]. Similarly, DKK1 promotes LRP6 internalization and degradation when forming a ternary complex with Kremen and LRP6, effectively inhibiting Wnt signaling [
61].
LGR6, considered a stem cell marker in various normal tissues, is associated with tissue development, regeneration, and repair. It has also been linked to the initiation and progression of certain cancers [
62]. Germline mutations in FGFR2 have been described in medulloblastoma [
63]. NXN (nucleoredoxin), a redox regulator of disheveled proteins, modulates WNT signaling, and its knockout in neuroblastoma cells affects self-renewal [
64]. Canonical Wnt signaling safeguards naïve pluripotency during embryogenesis, with TCF7L1 playing a repressive role during primitive endoderm induction [
65]. STK3 (alias MST-2) functions within the Hippo pathway during embryogenesis [
66] and interacts with WNT and Notch pathways in liver carcinogenesis [
67].
YAP and TAZ, transcriptional co-activators, are essential regulators of organ size and tissue homeostasis. Their dysregulation contributes to cancer, with alternative Wnt signaling pathways (Wnt5a/b and Wnt3a) activating YAP/TAZ independently of canonical β-catenin signaling. YAP/TAZ mediate alternative Wnt signaling effects, including antagonism of β-catenin-dependent pathways [
68]. Notably, the maintenance of undifferentiated embryonic stem cells by β-catenin is inversely correlated with YAP/TAZ activity [
69].
5. Conclusions
This study highlights the significant role of the transcription factor LEF1 and its associated gene targets in the Wnt signaling pathway across two embryonic tumor types, medulloblastoma and hepatoblastoma. Our findings provide evidence for a conserved transcriptional program regulated by LEF1, which is particularly enriched in WNT-subtype medulloblastoma and activated in tumor cells from hepatoblastoma. The identification of a 13-gene WNT-LEF1 signature underscores the utility of transcriptome-based analyses for distinguishing tumor subtypes and understanding underlying oncogenic mechanisms.
However, it is important to note that these conclusions are based on transcriptomic data. While these findings shed light on the molecular landscape of Wnt signaling in these tumors, further experimental studies are required to validate the functional roles of the identified genes. Investigations at the protein level, including proteomics and functional assays, will be essential to confirm the activity of these pathways and their impact on tumor behavior. Additionally, exploring post-transcriptional and epigenetic mechanisms could provide a more comprehensive understanding of WNT-LEF1 pathway regulation.
The results presented here emphasize the need for integrative, multi-omics approaches to validate and extend these findings. Future studies should aim to bridge the gap between transcriptional data and functional outcomes, potentially paving the way for novel therapeutic targets and biomarkers for these pediatric tumors. This section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on
Preprints.org.
Author Contributions
J.M.-G. and R.F designed the study. C.D. analyzed and interpreted the data and wrote the manuscript. C.D. performed most of the experiments, with contributions from Y.F., J.B-M and C.M. P.P. contributed to manuscript correction and data analysis. All authors have read and agreed to the published version of the manuscript.
Funding
P.P. and J.M-G. received the funding for this study from MEAE AMBASS FRANCE AU PEROU FSPI—S-AC23007, Filière Santé Maladie Rare du Foie de l’Adulte et de l’Enfant.
Data Availability Statement
Acknowledgments
We thank all the authors of the various works involving the experimental data used during this study and shared on opensource sites such as Gene Expression Omnibus and also the OpenPBTA initiative with access to pediatric cbioportal allowing a better understanding of omics for childhood brain tumors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sharma D, Subbarao G, Saxena R. Hepatoblastoma. Semin Diagn Pathol. 2017 Mar;34(2):192-200. Epub 2016 Dec 23. [CrossRef] [PubMed]
- Cotter JA, Hawkins C. Medulloblastoma: WHO 2021 and Beyond. Pediatr Dev Pathol. 2022 Jan-Feb;25(1):23-33. [CrossRef] [PubMed]
- Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, Zhou Z, Shu G, Yin G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. 2022 Jan 3;7(1):3. [CrossRef] [PubMed] [PubMed Central]
- He S, Tang S. WNT/β-catenin signaling in the development of liver cancers. Biomed Pharmacother. 2020 Dec;132:110851. Epub 2020 Oct 17. [CrossRef] [PubMed]
- Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat Rev. 2018 Jan;62:50-60. Epub 2017 Nov 13. [CrossRef] [PubMed] [PubMed Central]
- Crippa S, Ancey PB, Vazquez J, Angelino P, Rougemont AL, Guettier C, Zoete V, Delorenzi M, Michielin O, Meylan E. Mutant CTNNB1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol Med. 2017 Nov;9(11):1589-1604. [CrossRef] [PubMed] [PubMed Central]
- Chiang J, Moreira DC, Pytel NJ, Liu YC, Blackburn PR, Shi Z, Cardenas M, Wheeler DA, Furtado LV. A CTNNB1-altered medulloblastoma shows the immunophenotypic, DNA methylation and transcriptomic profiles of SHH-activated, and not WNT-activated, medulloblastoma. Neuropathol Appl Neurobiol. 2022 Aug;48(5):e12815. Epub 2022 Mar 30. [CrossRef] [PubMed] [PubMed Central]
- Staal FJ, Clevers H. Tcf/Lef transcription factors during T-cell development: unique and overlapping functions. Hematol J. 2000;1(1):3-6. [CrossRef] [PubMed]
- Gao L, Cui W, Kelting S, Woodroof J, Li H, Li L, Zhang D. Canonical and non-canonical Wnt signal pathway in classic Hodgkin lymphoma and the prognostic significance of LEF1, β-catenin, FZD6 and Wnt5a/b. Am J Blood Res. 2022 Aug 15;12(4):136-143. PMCID: PMC9490108. [PubMed]
- Katoh, M. Multi layered prevention and treatment of chronic inflammation, organ fibrosis and cancer associated with canonical WNT/β catenin signaling activation (Review). Int J Mol Med. 2018 Aug;42(2):713-725. Epub 2018 May 17. [CrossRef] [PubMed] [PubMed Central]
- Fakhr E, Zare F, Azadmanesh K, Teimoori-Toolabi L. LEF1 silencing sensitizes colorectal cancer cells to oxaliplatin, 5-FU, and irinotecan. Biomed Pharmacother. 2021 Nov;143:112091. Epub 2021 Aug 30. [CrossRef] [PubMed]
- Blazquez R, Rietkötter E, Wenske B, Wlochowitz D, Sparrer D, Vollmer E, Müller G, Seegerer J, Sun X, Dettmer K, Barrantes-Freer A, Stange L, Utpatel K, Bleckmann A, Treiber H, Bohnenberger H, Lenz C, Schulz M, Reimelt C, Hackl C, Grade M, Büyüktas D, Siam L, Balkenhol M, Stadelmann C, Kube D, Krahn MP, Proescholdt MA, Riemenschneider MJ, Evert M, Oefner PJ, Klein CA, Hanisch UK, Binder C, Pukrop T. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int J Cancer. 2020 Jun 1;146(11):3170-3183. Epub 2019 Nov 11. [CrossRef] [PubMed]
- Robinson, G., Parker, M., Kranenburg, T. A., Lu, C., Chen, X., Ding, L., Phoenix, T. N., Hedlund, E., Wei, L., Zhu, X., Chalhoub, N., Baker, S. J., Huether, R., Kriwacki, R., Curley, N., Thiruvenkatam, R., Wang, J., Wu, G., Rusch, M., Hong, X., Becksfort, J., Gupta, P., Ma, J., Easton, J., Vadodaria, B., Onar-Thomas, A., Lin, T., Li, S., Pounds, S., Paugh, S., Zhao, D., Kawauchi, D., Roussel, M. F., Finkelstein, D., Ellison, D. W., Lau, C. C., Bouffet, E., Hassall, T., Gururangan, S., Cohn, R., Fulton, R. S., Fulton, L. L., Dooling, D. J., Ochoa, K., Gajjar, A., Mardis, E. R., Wilson, R. K., Downing, J. R., Zhang, J., and Gilbertson, R. J. (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488, 43–48.
- Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., Marshall, K. A., Phillippy, K. H., Sherman, P. M., Holko, M., Yefanov, A., Lee, H., Zhang, N., Robertson, C. L., Serova, N., Davis, S., and Soboleva, A. (2013) NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 41, D991-995.
- Davis, S. and Meltzer, P. S. (2007) GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847.
- Lambert, S. R., Witt, H., Hovestadt, V., Zucknick, M., Kool, M., Pearson, D. M., Korshunov, A., Ryzhova, M., Ichimura, K., Jabado, N., Fontebasso, A. M., Lichter, P., Pfister, S. M., Collins, V. P., and Jones, D. T. W. (2013) Differential expression and methylation of brain developmental genes define location-specific subsets of pilocytic astrocytoma. Acta Neuropathol 126, 291–301.
- Riemondy, K. A., Venkataraman, S., Willard, N., Nellan, A., Sanford, B., Griesinger, A. M., Amani, V., Mitra, S., Hankinson, T. C., Handler, M. H., Sill, M., Ocasio, J., Weir, S. J., Malawsky, D. S., Gershon, T. R., Garancher, A., Wechsler-Reya, R. J., Hesselberth, J. R., Foreman, N. K., Donson, A. M., and Vibhakar, R. (2022) Neoplastic and immune single-cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro Oncol 24, 273–286.
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., and Satija, R. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420.
- Bondoc, A., Glaser, K., Jin, K., Lake, C., Cairo, S., Geller, J., Tiao, G., and Aronow, B. (2021) Identification of distinct tumor cell populations and key genetic mechanisms through single cell sequencing in hepatoblastoma. Commun Biol 4, 1049.
- Zappia, L., Lun, A., Kamm, J., and Cannoodt, R. (2024) zellkonverter: Conversion Between scRNA-seq Objects.
- Harikumar, A. and Meshorer, E. (2015) Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO Rep 16, 1609–1619.
- Leinonen, R., Sugawara, H., Shumway, M., and on behalf of the International Nucleotide Sequence Database Collaboration. (2011) The Sequence Read Archive. Nucleic Acids Research 39, D19–D21.
- Shapiro, J. A., Gaonkar, K. S., Spielman, S. J., Savonen, C. L., Bethell, C. J., Jin, R., Rathi, K. S., Zhu, Y., Egolf, L. E., Farrow, B. K., Miller, D. P., Yang, Y., Koganti, T., Noureen, N., Koptyra, M. P., Duong, N., Santi, M., Kim, J., Robins, S., Storm, P. B., Mack, S. C., Lilly, J. V., Xie, H. M., Jain, P., Raman, P., Rood, B. R., Lulla, R. R., Nazarian, J., Kraya, A. A., Vaksman, Z., Heath, A. P., Kline, C., Scolaro, L., Viaene, A. N., Huang, X., Way, G. P., Foltz, S. M., Zhang, B., Poetsch, A. R., Mueller, S., Ennis, B. M., Prados, M., Diskin, S. J., Zheng, S., Guo, Y., Kannan, S., Waanders, A. J., Margol, A. S., Kim, M. C., Hanson, D., Van Kuren, N., Wong, J., Kaufman, R. S., Coleman, N., Blackden, C., Cole, K. A., Mason, J. L., Madsen, P. J., Koschmann, C. J., Stewart, D. R., Wafula, E., Brown, M. A., Resnick, A. C., Greene, C. S., Rokita, J. L., Taroni, J. N., Children’s Brain Tumor Network, and Pacific Pediatric Neuro-Oncology Consortium. (2023) OpenPBTA: The Open Pediatric Brain Tumor Atlas. Cell Genom 3, 100340.
- Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E., and Storey, J. D. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883.
- Zhao, Y., Wong, L., and Goh, W. W. B. (2020) How to do quantile normalization correctly for gene expression data analyses. Sci Rep 10, 15534.
- Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., and Smyth, G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47.
- Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L., Fu, X., Liu, S., Bo, X., and Yu, G. (2021) clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation 2, 100141.
- Xu, S., Hu, E., Cai, Y., Xie, Z., Luo, X., Zhan, L., Tang, W., Wang, Q., Liu, B., Wang, R., Xie, W., Wu, T., Xie, L., and Yu, G. (2024) Using clusterProfiler to characterize multiomics data. Nat Protoc 19, 3292–3320.
- Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., Harris, M. A., Hill, D. P., Issel-Tarver, L., Kasarskis, A., Lewis, S., Matese, J. C., Richardson, J. E., Ringwald, M., Rubin, G. M., and Sherlock, G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29.
- Landt, S. G., Marinov, G. K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., Bernstein, B. E., Bickel, P., Brown, J. B., Cayting, P., Chen, Y., DeSalvo, G., Epstein, C., Fisher-Aylor, K. I., Euskirchen, G., Gerstein, M., Gertz, J., Hartemink, A. J., Hoffman, M. M., Iyer, V. R., Jung, Y. L., Karmakar, S., Kellis, M., Kharchenko, P. V., Li, Q., Liu, T., Liu, X. S., Ma, L., Milosavljevic, A., Myers, R. M., Park, P. J., Pazin, M. J., Perry, M. D., Raha, D., Reddy, T. E., Rozowsky, J., Shoresh, N., Sidow, A., Slattery, M., Stamatoyannopoulos, J. A., Tolstorukov, M. Y., White, K. P., Xi, S., Farnham, P. J., Lieb, J. D., Wold, B. J., and Snyder, M. (2012) ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 22, 1813–1831.
- Bolger, A. M., Lohse, M., and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120.
- Langmead, B. and Salzberg, S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., and 1000 Genome Project Data Processing Subgroup. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079.
- Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P., and Marth, G. T. (2011) BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W., and Liu, X. S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137.
- Feng, J., Liu, T., Qin, B., Zhang, Y., and Liu, X. S. (2012) Identifying ChIP-seq enrichment using MACS. Nat Protoc 7, 1728–1740.
- Liang, K. and Keleş, S. (2012) Normalization of ChIP-seq data with control. BMC Bioinformatics 13, 199.
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., Cheng, J. X., Murre, C., Singh, H., and Glass, C. K. (2010) Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589.
- Frankish, A., Diekhans, M., Jungreis, I., Lagarde, J., Loveland, J. E., Mudge, J. M., Sisu, C., Wright, J. C., Armstrong, J., Barnes, I., Berry, A., Bignell, A., Boix, C., Carbonell Sala, S., Cunningham, F., Di Domenico, T., Donaldson, S., Fiddes, I. T., García Girón, C., Gonzalez, J. M., Grego, T., Hardy, M., Hourlier, T., Howe, K. L., Hunt, T., Izuogu, O. G., Johnson, R., Martin, F. J., Martínez, L., Mohanan, S., Muir, P., Navarro, F. C. P., Parker, A., Pei, B., Pozo, F., Riera, F. C., Ruffier, M., Schmitt, B. M., Stapleton, E., Suner, M.-M., Sycheva, I., Uszczynska-Ratajczak, B., Wolf, M. Y., Xu, J., Yang, Y. T., Yates, A., Zerbino, D., Zhang, Y., Choudhary, J. S., Gerstein, M., Guigó, R., Hubbard, T. J. P., Kellis, M., Paten, B., Tress, M. L., and Flicek, P. (2021) GENCODE 2021. Nucleic Acids Res 49, D916–D923.
- Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A., and Manke, T. (2014) deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187-191.
- Thiele, C. and Hirschfeld, G. (2021) cutpointr: Improved Estimation and Validation of Optimal Cutpoints in R. J. Stat. Soft. 98.
- Kuhn, M. (2008) Building Predictive Models in R Using the caret Package. J. Stat. Soft. 28.
- Tay, J. K., Narasimhan, B., and Hastie, T. (2023) Elastic Net Regularization Paths for All Generalized Linear Models. J. Stat. Soft. 106.
- Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y., Albu, M., Chen, X., Taipale, J., Hughes, T. R., and Weirauch, M. T. (2018) The Human Transcription Factors. Cell 172, 650–665.
- Estarás, C., Benner, C., and Jones, K. A. (2015) SMADs and YAP compete to control elongation of β-catenin:LEF-1-recruited RNAPII during hESC differentiation. Mol Cell 58, 780–793.
- Geng, Z., Wafula, E., Corbett, R. J., Zhang, Y., Jin, R., Gaonkar, K. S., Shukla, S., Rathi, K. S., Hill, D., Lahiri, A., Miller, D. P., Sickler, A., Keith, K., Blackden, C., Chroni, A., Brown, M. A., Kraya, A. A., Koschmann, C. J., Aldape, K., Huang, X., Rood, B. R., Mason, J. L., Trooskin, G. R., Abdullaev, Z., Wang, P., Zhu, Y., Farrow, B. K., Farrel, A., Dybas, J. M., Zhong, C., Van Kuren, N., Zhang, B., Santi, M., Phul, S., Chinwalla, A. T., Resnick, A. C., Diskin, S. J., Tasian, S., Stefankiewicz, S., Maris, J. M., Ennis, B. M., Lueder, M. R., Naqvi, A. S., Coleman, N., Ma, W., Taylor, D., and Rokita, J. L. (2024) The Open Pediatric Cancer Project.
- Cairo, S., Armengol, C., De Reyniès, A., Wei, Y., Thomas, E., Renard, C.-A., Goga, A., Balakrishnan, A., Semeraro, M., Gresh, L., Pontoglio, M., Strick-Marchand, H., Levillayer, F., Nouet, Y., Rickman, D., Gauthier, F., Branchereau, S., Brugières, L., Laithier, V., Bouvier, R., Boman, F., Basso, G., Michiels, J.-F., Hofman, P., Arbez-Gindre, F., Jouan, H., Rousselet-Chapeau, M.-C., Berrebi, D., Marcellin, L., Plenat, F., Zachar, D., Joubert, M., Selves, J., Pasquier, D., Bioulac-Sage, P., Grotzer, M., Childs, M., Fabre, M., and Buendia, M.-A. (2008) Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 14, 471–484.
- Ramaswamy, V., Remke, M., Bouffet, E., Bailey, S., Clifford, S. C., Doz, F., Kool, M., Dufour, C., Vassal, G., Milde, T., Witt, O., von Hoff, K., Pietsch, T., Northcott, P. A., Gajjar, A., Robinson, G. W., Padovani, L., André, N., Massimino, M., Pizer, B., Packer, R., Rutkowski, S., Pfister, S. M., Taylor, M. D., and Pomeroy, S. L. (2016) Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 131, 821–831.
- Wang, D., Gong, J., Zhang, H., Liu, Y., Sun, N., Hao, X., and Mu, K. (2022) Immunohistochemical staining of LEF-1 is a useful marker for distinguishing WNT-activated medulloblastomas. Diagn Pathol 17, 69.
- Aboubakr, O., Métais, A., Doz, F., Saffroy, R., Masliah-Planchon, J., Hasty, L., Beccaria, K., Ayrault, O., Dufour, C., Varlet, P., and Tauziède-Espariat, A. (2024) LEF-1 immunohistochemistry, a better diagnostic biomarker than β-catenin for medulloblastoma, WNT-activated subtyping. J Neuropathol Exp Neurol 83, 136–138.
- Ye, S., Zhang, T., Tong, C., Zhou, X., He, K., Ban, Q., Liu, D., and Ying, Q.-L. (2017) Depletion of Tcf3 and Lef1 maintains mouse embryonic stem cell self-renewal. Biol Open 6, 511–517.
- Kobayashi, W. and Ozawa, M. (2013) The transcription factor LEF-1 induces an epithelial–mesenchymal transition in MDCK cells independent of β-catenin. Biochemical and Biophysical Research Communications 442, 133–138.
- Zirkel, A., Lederer, M., Stöhr, N., Pazaitis, N., and Hüttelmaier, S. (2013) IGF2BP1 promotes mesenchymal cell properties and migration of tumor-derived cells by enhancing the expression of LEF1 and SNAI2 (SLUG). Nucleic Acids Research 41, 6618–6636.
- Liang, J., Li, X., Li, Y., Wei, J., Daniels, G., Zhong, X., Wang, J., Sfanos, K., Melamed, J., Zhao, J., and Lee, P. (2015) LEF1 targeting EMT in prostate cancer invasion is mediated by miR-181a. Am J Cancer Res 5, 1124–1132.
- Eberherr, C., Beck, A., Vokuhl, C., Becker, K., Häberle, B., Von Schweinitz, D., and Kappler, R. (2019) Targeting excessive MYCN expression using MLN8237 and JQ1 impairs the growth of hepatoblastoma cells. Int J Oncol 54, 1853–1863.
- Bourdeaut, F., Grison, C., Maurage, C.-A., Laquerriere, A., Vasiljevic, A., Delisle, M.-B., Michalak, S., Figarella-Branger, D., Doz, F., Richer, W., Pierron, G., Miquel, C., Delattre, O., and Couturier, J. (2013) MYC and MYCN amplification can be reliably assessed by aCGH in medulloblastoma. Cancer Genet 206, 124–129.
- de Lau, W., Peng, W. C., Gros, P., and Clevers, H. (2014) The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev 28, 305–316.
- Bell, I., Khan, H., Stutt, N., Horn, M., Hydzik, T., Lum, W., Rea, V., Clapham, E., Hoeg, L., and Van Raay, T. J. (2024) Nkd1 functions downstream of Axin2 to attenuate Wnt signaling. Mol Biol Cell 35, ar93.
- Fatima, S., Lee, N. P., Tsang, F. H., Kolligs, F. T., Ng, I. O. L., Poon, R. T. P., Fan, S. T., and Luk, J. M. (2012) Dickkopf 4 (DKK4) acts on Wnt/β-catenin pathway by influencing β-catenin in hepatocellular carcinoma. Oncogene 31, 4233–4244.
- Li, Y., Lu, W., King, T. D., Liu, C.-C., Bijur, G. N., and Bu, G. (2010) Dkk1 stabilizes Wnt co-receptor LRP6: implication for Wnt ligand-induced LRP6 down-regulation. PLoS One 5, e11014.
- Feng, Q., Li, S., Ma, H.-M., Yang, W.-T., and Zheng, P.-S. (2021) LGR6 activates the Wnt/β-catenin signaling pathway and forms a β-catenin/TCF7L2/LGR6 feedback loop in LGR6high cervical cancer stem cells. Oncogene 40, 6103–6114.
- Bourdeaut, F., Miquel, C., Di Rocco, F., Grison, C., Richer, W., Brugieres, L., Pierron, G., James, S., Baujat, G., Delattre, O., and Collet, C. (2013) Germline mutations in FGF receptors and medulloblastomas. American J of Med Genetics Pt A 161, 382–385.
- Valek, L. and Tegeder, I. (2021) Nucleoredoxin Knockdown in SH-SY5Y Cells Promotes Cell Renewal. Antioxidants (Basel) 10, 449.
- Athanasouli, P., Balli, M., De Jaime-Soguero, A., Boel, A., Papanikolaou, S., van der Veer, B. K., Janiszewski, A., Vanhessche, T., Francis, A., El Laithy, Y., Nigro, A. L., Aulicino, F., Koh, K. P., Pasque, V., Cosma, M. P., Verfaillie, C., Zwijsen, A., Heindryckx, B., Nikolaou, C., and Lluis, F. (2023) The Wnt/TCF7L1 transcriptional repressor axis drives primitive endoderm formation by antagonizing naive and formative pluripotency. Nat Commun 14, 1210.
- Li, P., Chen, Y., Mak, K. K., Wong, C. K., Wang, C. C., and Yuan, P. (2013) Functional role of Mst1/Mst2 in embryonic stem cell differentiation. PLoS One 8, e79867.
- Kim, W., Khan, S. K., Gvozdenovic-Jeremic, J., Kim, Y., Dahlman, J., Kim, H., Park, O., Ishitani, T., Jho, E.-H., Gao, B., and Yang, Y. (2017) Hippo signaling interactions with Wnt/β-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 127, 137–152.
- Park, H. W., Kim, Y. C., Yu, B., Moroishi, T., Mo, J.-S., Plouffe, S. W., Meng, Z., Lin, K. C., Yu, F.-X., Alexander, C. M., Wang, C.-Y., and Guan, K.-L. (2015) Alternative Wnt Signaling Activates YAP/TAZ. Cell 162, 780–794.
- Azzolin, L., Panciera, T., Soligo, S., Enzo, E., Bicciato, S., Dupont, S., Bresolin, S., Frasson, C., Basso, G., Guzzardo, V., Fassina, A., Cordenonsi, M., and Piccolo, S. (2014) YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170.
- Morfouace, M., Cheepala, S., Jackson, S., Fukuda, Y., Patel, Y. T., Fatima, S., Kawauchi, D., Shelat, A. A., Stewart, C. F., Sorrentino, B. P., Schuetz, J. D., and Roussel, M. F. (2015) ABCG2 Transporter Expression Impacts Group 3 Medulloblastoma Response to Chemotherapy. Cancer Res 75, 3879–3889.
Figure 1.
Common transcriptional regulation between hepatoblastoma and medulloblastoma tumors: cross-combat normalization between GSE37418, GSE44971, and GSE104766 datasets. (a) Principal component analysis (PCA) performed on the 1,259 differentially expressed genes (DEGs) regulated between tumor samples (HB: hepatoblastoma + MB: medulloblastoma) and control samples (normal cerebellum + normal liver). (b) Unsupervised clustering (Euclidean distances) based on the common HB-MB signature (1,259 DEGs). (c) Volcano plot highlighting the transcription factors regulated between tumors (HB + MB) and normal tissues (cerebellum + liver).
Figure 1.
Common transcriptional regulation between hepatoblastoma and medulloblastoma tumors: cross-combat normalization between GSE37418, GSE44971, and GSE104766 datasets. (a) Principal component analysis (PCA) performed on the 1,259 differentially expressed genes (DEGs) regulated between tumor samples (HB: hepatoblastoma + MB: medulloblastoma) and control samples (normal cerebellum + normal liver). (b) Unsupervised clustering (Euclidean distances) based on the common HB-MB signature (1,259 DEGs). (c) Volcano plot highlighting the transcription factors regulated between tumors (HB + MB) and normal tissues (cerebellum + liver).
Figure 2.
Embryonic program of LEF1 genome binding occupancy after WNT3A stimulation: genomic heatmap illustrating the enrichment of genomic intervals (HG38) for LEF1 binding in H1 human embryonic stem cell chromatin after WNT3A stimulation. From left to right, respective enrichments are shown for ChIP-seq of H1 input chromatin, LEF1 in unstimulated H1 cells, H3 histone lysine 27 trimethylation (repressive mark), H3 histone lysine 27 acetylation (active mark), and LEF1 in WNT3A-stimulated H1 cells. (TSS: transcription start site, TTS: transcription termination site).
Figure 2.
Embryonic program of LEF1 genome binding occupancy after WNT3A stimulation: genomic heatmap illustrating the enrichment of genomic intervals (HG38) for LEF1 binding in H1 human embryonic stem cell chromatin after WNT3A stimulation. From left to right, respective enrichments are shown for ChIP-seq of H1 input chromatin, LEF1 in unstimulated H1 cells, H3 histone lysine 27 trimethylation (repressive mark), H3 histone lysine 27 acetylation (active mark), and LEF1 in WNT3A-stimulated H1 cells. (TSS: transcription start site, TTS: transcription termination site).
Figure 3.
LEF1 embryonic binding program predicts the WNT subtype in the medulloblastoma transcriptome: integration of the LEF1 binding program in the MB-PBTA transcriptome cohort. (a) Expression of LEF1 in the medulloblastoma transcriptome of the PBTA cohort stratified by medulloblastoma subtypes: WNT, SHH, G3, G4. (b) Volcano plot of differentially expressed genes (with embryonic LEF1 binding) between the WNT-MB subtype and other MB samples. (c) Principal component analysis (PCA) performed on the 141 LEF1 embryonic targets overexpressed in the WNT-MB subtype. (d) Unsupervised clustering (Euclidean distances) based on the expression of the 141 LEF1 embryonic targets overexpressed in the WNT-MB subtype.
Figure 3.
LEF1 embryonic binding program predicts the WNT subtype in the medulloblastoma transcriptome: integration of the LEF1 binding program in the MB-PBTA transcriptome cohort. (a) Expression of LEF1 in the medulloblastoma transcriptome of the PBTA cohort stratified by medulloblastoma subtypes: WNT, SHH, G3, G4. (b) Volcano plot of differentially expressed genes (with embryonic LEF1 binding) between the WNT-MB subtype and other MB samples. (c) Principal component analysis (PCA) performed on the 141 LEF1 embryonic targets overexpressed in the WNT-MB subtype. (d) Unsupervised clustering (Euclidean distances) based on the expression of the 141 LEF1 embryonic targets overexpressed in the WNT-MB subtype.
Figure 4.
Heterogeneity of LEF1-dependent embryonic promoters for targets activated during WNT-medulloblastoma: integration into the MB-PBTA cohort. (a) Scatterplot of the integrative analysis representing ChIP-seq peak distances from TSS versus log2 fold changes in the WNT-MB transcriptome. Dot sizes represent peak scores from the MACS2 algorithm for LEF1-H1-WNT3A ChIP-seq data. (b) Promoter visualization for the LEF1 and LEF1-AS loci. (c) Promoter visualization for the DKK4 locus. (d) Promoter visualization for the RNF43 locus. (e) Promoter visualization for the MYCN locus.
Figure 4.
Heterogeneity of LEF1-dependent embryonic promoters for targets activated during WNT-medulloblastoma: integration into the MB-PBTA cohort. (a) Scatterplot of the integrative analysis representing ChIP-seq peak distances from TSS versus log2 fold changes in the WNT-MB transcriptome. Dot sizes represent peak scores from the MACS2 algorithm for LEF1-H1-WNT3A ChIP-seq data. (b) Promoter visualization for the LEF1 and LEF1-AS loci. (c) Promoter visualization for the DKK4 locus. (d) Promoter visualization for the RNF43 locus. (e) Promoter visualization for the MYCN locus.
Figure 5.
WNT signaling LEF1-dependent signature in medulloblastoma: MB-PBTA cohort. (a) Functional enrichment network based on LEF1 targets activated during the WNT-medulloblastoma subtype using the Gene Ontology Biological Process (GO-BP) database. (b) Unsupervised clustering (Euclidean distances) of the WNT-program LEF1-dependent genes in medulloblastoma tumors. (c) Principal component analysis (PCA) of the WNT-program LEF1-dependent genes in medulloblastoma tumors. (d) Univariate binomial regression analysis of the 13 WNT signaling target genes of LEF1 during medulloblastoma (outcome: WNT subtype).
Figure 5.
WNT signaling LEF1-dependent signature in medulloblastoma: MB-PBTA cohort. (a) Functional enrichment network based on LEF1 targets activated during the WNT-medulloblastoma subtype using the Gene Ontology Biological Process (GO-BP) database. (b) Unsupervised clustering (Euclidean distances) of the WNT-program LEF1-dependent genes in medulloblastoma tumors. (c) Principal component analysis (PCA) of the WNT-program LEF1-dependent genes in medulloblastoma tumors. (d) Univariate binomial regression analysis of the 13 WNT signaling target genes of LEF1 during medulloblastoma (outcome: WNT subtype).
Figure 6.
Validation of the WNT-LEF1 signature in independent medulloblastoma cohorts: GSE37418 and scRNA-seq GSE155446 datasets. (a) Unsupervised clustering (Euclidean distances) based on the expression of the WNT-LEF1-dependent program (GSE37418). (b) Principal component analysis (PCA) of the WNT-LEF1-dependent program (GSE37418). (c) ElasticNet tuning of lambda and alpha parameters to predict the WNT subgroup using the WNT-LEF1-dependent program. (d) Barplot of ElasticNet coefficients predicting the WNT subtype in the GSE37418 cohort. (e) WNT-MB subtypes in the single-cell transcriptome dataset GSE155446 (28 MB tumor samples). (f) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE155446.
Figure 6.
Validation of the WNT-LEF1 signature in independent medulloblastoma cohorts: GSE37418 and scRNA-seq GSE155446 datasets. (a) Unsupervised clustering (Euclidean distances) based on the expression of the WNT-LEF1-dependent program (GSE37418). (b) Principal component analysis (PCA) of the WNT-LEF1-dependent program (GSE37418). (c) ElasticNet tuning of lambda and alpha parameters to predict the WNT subgroup using the WNT-LEF1-dependent program. (d) Barplot of ElasticNet coefficients predicting the WNT subtype in the GSE37418 cohort. (e) WNT-MB subtypes in the single-cell transcriptome dataset GSE155446 (28 MB tumor samples). (f) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE155446.
Figure 7.
Validation of the WNT-LEF1 signature in an independent hepatoblastoma cohort: GSE131329 and scRNA-seq GSE180655 datasets. (a) Unsupervised clustering (Euclidean distances) based on the expression of the WNT-LEF1 program (GSE131329). (b) Principal component analysis (PCA) of the WNT-LEF1 program (GSE131329). (c) UMAP dimensionality reduction showing sample identities in the scRNA-seq dataset GSE180655. (d) UMAP dimensionality reduction showing cell type identities in the scRNA-seq dataset GSE180655. (e) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE180655, stratified by sample types (background: adjacent normal liver; pdx: patient-derived xenotransplantation model; tumor: HB tumor samples). (f) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE180655, stratified by cell types.
Figure 7.
Validation of the WNT-LEF1 signature in an independent hepatoblastoma cohort: GSE131329 and scRNA-seq GSE180655 datasets. (a) Unsupervised clustering (Euclidean distances) based on the expression of the WNT-LEF1 program (GSE131329). (b) Principal component analysis (PCA) of the WNT-LEF1 program (GSE131329). (c) UMAP dimensionality reduction showing sample identities in the scRNA-seq dataset GSE180655. (d) UMAP dimensionality reduction showing cell type identities in the scRNA-seq dataset GSE180655. (e) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE180655, stratified by sample types (background: adjacent normal liver; pdx: patient-derived xenotransplantation model; tumor: HB tumor samples). (f) WNT-LEF1-dependent score in the single-cell RNA-seq dataset GSE180655, stratified by cell types.
Table 1.
WNT-LEF1 score stratification for PBTA medulloblastoma cohort.
Table 1.
WNT-LEF1 score stratification for PBTA medulloblastoma cohort.
| Variable |
Level |
HIGH (n= 21) |
low (n=233) |
Total (n=254) |
p-value |
| SEX |
Male |
13 (61.9) |
142 (60.9) |
155 (61.0) |
|
| |
Female |
8 (38.1) |
91 (39.1) |
99 (39.0) |
1.00000 |
| TUMOR TYPE |
primary |
20 (95.2) |
135 (57.9) |
155 (61.0) |
|
| |
progression |
0 (0.0) |
3 (1.3) |
3 (1.2) |
|
| |
metastatic |
1 (4.8) |
83 (35.6) |
84 (33.1) |
|
| |
recurrence |
0 (0.0) |
11 (4.7) |
11 (4.3) |
|
| |
Deceased |
0 (0.0) |
1 (0.4) |
1 (0.4) |
0.02348 |
| group |
WNT |
21 (100.0) |
0 (0.0) |
21 (8.3) |
|
| |
G3 |
0 (0.0) |
60 (25.8) |
60 (23.6) |
|
| |
G4 |
0 (0.0) |
99 (42.5) |
99 (39.0) |
|
| |
SHH |
0 (0.0) |
74 (31.8) |
74 (29.1) |
< 1e-04 |
| CNS_REGION |
Ventricles |
5 (23.8) |
12 (5.2) |
17 (6.7) |
|
| |
Mixed |
3 (14.3) |
63 (27.2) |
66 (26.1) |
|
| |
Posterior fossa |
13 (61.9) |
152 (65.5) |
165 (65.2) |
|
| |
Spine |
0 (0.0) |
1 (0.4) |
1 (0.4) |
|
| |
Hemispheric |
0 (0.0) |
3 (1.3) |
3 (1.2) |
|
| |
Other |
0 (0.0) |
1 (0.4) |
1 (0.4) |
0.03963 |
| |
missing |
0 |
1 |
1 |
|
| EFS_STATUS |
1:Recurrence |
1 (4.8) |
36 (15.5) |
37 (14.6) |
|
| |
0:No Event |
19 (90.5) |
121 (51.9) |
140 (55.1) |
|
| |
1:Progressive - Metastatic |
1 (4.8) |
33 (14.2) |
34 (13.4) |
|
| |
1:Deceased - due to disease |
0 (0.0) |
7 (3.0) |
7 (2.8) |
|
| |
1:Recurrence - Metastatic |
0 (0.0) |
23 (9.9) |
23 (9.1) |
|
| |
1:Second Malignancy |
0 (0.0) |
6 (2.6) |
6 (2.4) |
|
| |
1:Progressive |
0 (0.0) |
7 (3.0) |
7 (2.8) |
0.06538 |
Table 2.
WNT-LEF1 score stratification for GSE37418 medulloblastoma validation cohort.
Table 2.
WNT-LEF1 score stratification for GSE37418 medulloblastoma validation cohort.
| Variable |
Level |
Low (n=66) |
Hight (n= 8) |
Total (n=74) |
p-value |
| gender |
Male |
51 (77.3) |
2 (25.0) |
53 (71.6) |
|
| |
Female |
15 (22.7) |
6 (75.0) |
21 (28.4) |
0.00732 |
| age_months |
mean (sd) |
98.4 (38.7) |
104.5 (17) |
99.1 (37) |
0.66172 |
| anapath |
CL |
43 (65.2) |
8 (100.0) |
51 (68.9) |
|
| |
DN |
6 (9.1) |
0 (0.0) |
6 (8.1) |
|
| |
AN |
17 (25.8) |
0 (0.0) |
17 (23.0) |
0.13231 |
| ethnie |
White |
38 (57.6) |
4 (50.0) |
42 (56.8) |
|
| |
Black |
5 (7.6) |
0 (0.0) |
5 (6.8) |
|
| |
Asian |
5 (7.6) |
1 (12.5) |
6 (8.1) |
|
| |
other |
2 (3.0) |
1 (12.5) |
3 (4.1) |
|
| |
Hispanic |
12 (18.2) |
1 (12.5) |
13 (17.6) |
|
| |
Asian Indian |
4 (6.1) |
0 (0.0) |
4 (5.4) |
|
| |
Pacific Islander |
0 (0.0) |
1 (12.5) |
1 (1.4) |
0.07854 |
| group |
G4 |
39 (59.1) |
0 (0.0) |
39 (52.7) |
|
| |
WNT |
0 (0.0) |
8 (100.0) |
8 (10.8) |
|
| |
SHH |
11 (16.7) |
0 (0.0) |
11 (14.9) |
|
| |
G3 |
16 (24.2) |
0 (0.0) |
16 (21.6) |
< 1e-04 |
Table 3.
Corhort description for hepatoblastoma validation cohort GSE131329.
Table 3.
Corhort description for hepatoblastoma validation cohort GSE131329.
| Variable |
Level |
tumor_tissue (n=53) |
Noncancerous liver tissue (n=14) |
Total (n=67) |
p-value |
| gender |
Female |
25 (47.2) |
8 (57.1) |
33 (49.3) |
|
| |
Male |
28 (52.8) |
6 (42.9) |
34 (50.7) |
0.716363 |
| Histological type |
Well_differentiated |
30 (56.6) |
14 (100.0) |
44 (65.7) |
|
| |
Other |
2 (3.8) |
0 (0.0) |
2 (3.0) |
|
| |
Poorly_differentiated |
21 (39.6) |
0 (0.0) |
21 (31.3) |
0.009797 |
| age_months |
mean (sd) |
27.2 (24.1) |
27.6 (26.4) |
27.3 (24.4) |
0.964822 |
| pretext_stage |
P3 |
18 (34.0) |
0 (0.0) |
18 (34.0) |
|
| |
P2 |
15 (28.3) |
0 (0.0) |
15 (28.3) |
|
| |
P4 |
11 (20.8) |
0 (0.0) |
11 (20.8) |
|
| |
P1 |
9 (17.0) |
0 (0.0) |
9 (17.0) |
NA |
| chic_risk stratification |
Standard |
31 (58.5) |
0 (0.0) |
31 (58.5) |
|
| |
High |
14 (26.4) |
0 (0.0) |
14 (26.4) |
|
| |
Intermediate |
8 (15.1) |
0 (0.0) |
8 (15.1) |
NA |
| ctnnbi_gene alteration |
Deletion |
23 (43.4) |
0 (0.0) |
23 (43.4) |
|
| |
Wild_Type |
14 (26.4) |
0 (0.0) |
14 (26.4) |
|
| |
Mutation (exon3) |
16 (30.2) |
0 (0.0) |
16 (30.2) |
NA |
| clinical_course |
Alive |
38 (71.7) |
0 (0.0) |
38 (71.7) |
|
| |
Dead |
15 (28.3) |
0 (0.0) |
15 (28.3) |
NA |
Table 4.
Cell type comparison for WNT-LEF1 score in single cell transcriptome of hepatoblastoma.
Table 4.
Cell type comparison for WNT-LEF1 score in single cell transcriptome of hepatoblastoma.
| Comparison |
Mean Groupe1 |
Mean Groupe2 |
p_value |
| Hepatocyte vs Tumor cell |
-1.088 |
0.450 |
0.00E+00 |
| Hepatic Stellate cell vs Tumor cell |
0.168 |
0.450 |
2.93E-07 |
| Endothelial cell vs Tumor cell |
-0.264 |
0.450 |
0.00E+00 |
| Kupffer cell vs Tumor cell |
-0.686 |
0.450 |
0.00E+00 |
| B cell vs Tumor cell |
-0.628 |
0.450 |
1.24E+05 |
| Proliferative hepatocyte vs Tumor cell |
-1.270 |
0.450 |
2.40E-24 |
| T cell_NK cell vs Tumor cell |
-0.652 |
0.450 |
9.86E-91 |
| Cholangiocyte vs Tumor cell |
-0.208 |
0.450 |
2.26E-07 |
| Lymphatic vs Tumor cell |
-0.416 |
0.450 |
7.62E+07 |
| T_NK cell vs Tumor cell |
-0.267 |
0.450 |
1.25E-59 |
| Neuronal cell vs Tumor cell |
0.375 |
0.450 |
4.13E-01 |
| Erythrocyte vs Tumor cells |
-0.619 |
0.450 |
4.20E-50 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).