
Submitted:
11 December 2024
Posted:
12 December 2024
You are already at the latest version
Abstract
This review describes mass spectrometry (MS)-based approaches for the absolute quantification of therapeutic monoclonal antibodies (mAbs), focusing on technical challenges in sample treatment and calibration. Therapeutic mAbs are crucial for treating cancer, inflammatory, infectious, and autoimmune diseases. We trace their development from hybridoma technology and the first murine mAbs in 1975 to today’s chimeric and fully human mAbs. With increasing commercial relevance, absolute quantification of mAbs, traceable to SI units, has attracted attention from science, industry, and national metrology institutes (NMIs). Quantification of proteotypic peptides after enzymatic digestion using liquid chromatography-tandem mass spectrometry (LC-MS/MS) has emerged as the most viable strategy, though methods targeting intact mAbs are still being explored. We review peptide-based quantification, focusing on critical experimental steps like denaturation, reduction, alkylation, choice of digestion enzyme, and selection of signature peptides. Challenges in amino acid analysis (AAA) for quantifying pure mAbs and peptide calibrators, along with software tools for targeted MS data analysis, are also discussed. Short explanations within each chapter provide newcomers an overview of the field’s challenges. Finally, we discuss prospects and limitations of developing standardized protocols and certified reference materials (CRMs) and suggest future applications of newer technologies for the absolute quantification of therapeutic antibodies.
Keywords:
monoclonal antibody
; therapeutic antibodies
; mass spectrometry
; liquid chromatography
; absolute quantification
; isotope labelling
; metrology
; traceability
; certified reference material
1. Historical Overview: Evolution of Recombinant mAbs
The development of hybridoma technology by Köhler and Milstein in 1975 provided the cornerstone for the generation of monoclonal antibodies. By fusing B lymphocytes from immunized animals with myeloma cells, stable cell lines for unlimited monoclonal antibody (mAb) production could be obtained for the first time [1]. Through their ability to bind with high selectivity and affinity to a wide range of targeting biomolecules, mAbs gained increasing attention for application in life science and medicine. The first murine mAb approved by the Food and Drug Administration (FDA) for therapeutic application on humans was Muromonab in 1986 [2]. Unfortunately, the murine nature of antibodies generated by hybridoma technology bore some disadvantages for their human therapeutic application. The immune system recognized the injected mAbs as foreign proteins, resulting in the production of antibodies against those, the so-called human anti-mouse antibody (HAMA) response [3]. Consequently, the administered antibodies were eliminated from the body, reducing the effectiveness of treatment or causing allergic reactions [4].
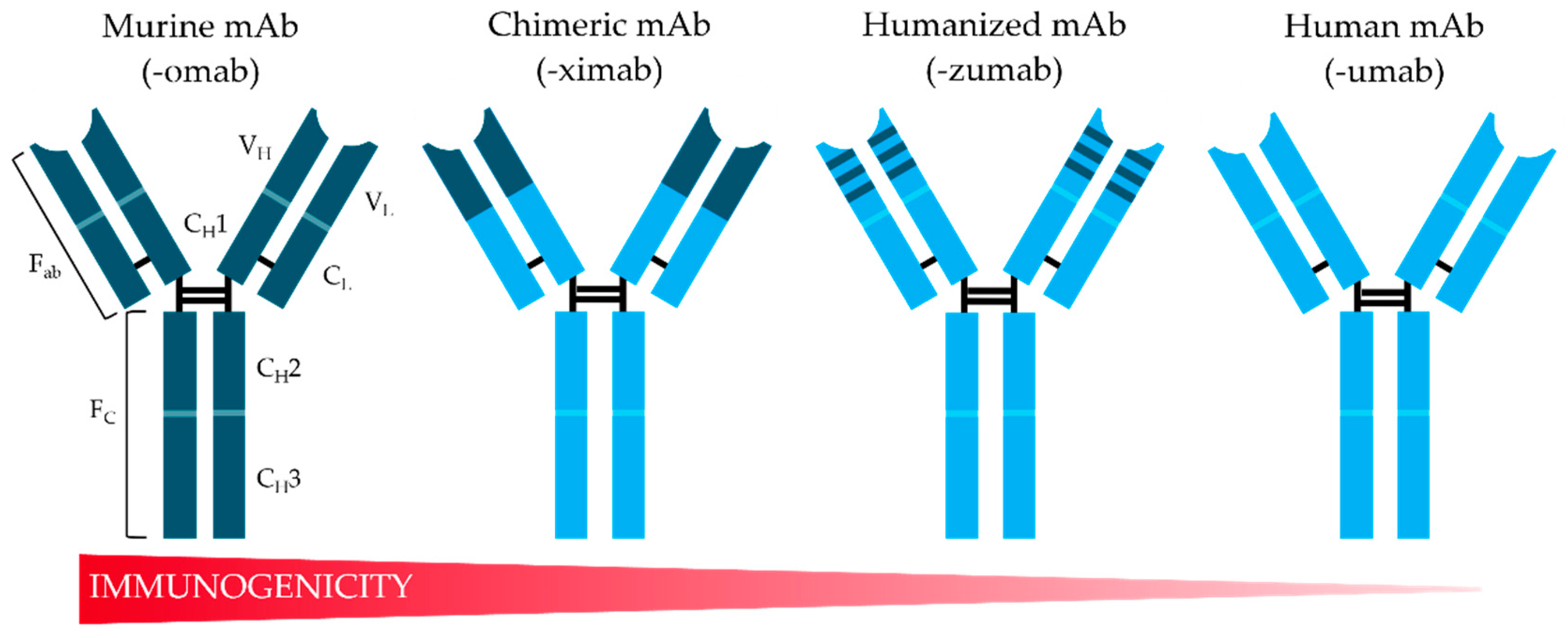
To overcome such limitations, the field of recombinant DNA technologies offers a wide variety of tools for structural modifications of murine mAbs to create more human-like antibodies and enable large-scale production using mammalian cell lines. Initial achievements have been made by combining a murine variable domain with a human Fc-region (Fragment crystallizable, see Figure 1) to generate chimeric antibodies [5] with reduced, but still observable immunogenicity in humans [6]. The first FDA-approved chimeric antibodies were Abciximab in 1994 [7] and Rituximab in 1997, a recombinant antibody produced in Chinese hamster ovary (CHO) cells [8]. Further developments enabled the reduction of murine parts in mAbs, such as complementarity-determining region (CDR) grafting, whereby it was possible to incorporate hypervariable loops from antigen-binding sites (Fab) of the murine antibody into a fully human antibody scaffold [9] (Figure 1). These humanized antibodies were even less immunogenic than chimeric mAbs, but in some cases showed reduced affinity to their target antigen [10]. The first FDA-approved humanized mAb was Daclizumab in 2016, an immunosuppressive agent used to reduce renal transplant rejection in patients [11]. Since then, various methods have been developed for the humanization of therapeutic antibodies to improve their properties and make them applicable to medical treatment [12].
Another notable step in the evolution of therapeutic antibodies was the development of antibody phage display technology [13], based on the previously established phage display method by Smith et al. in 1985 [14]. For this approach, a library of antibody gene fragments from immunized or non-immunized animals as well as humans are needed. Additionally, fully synthetic libraries were established, as reviewed recently by Zhang et al. 2023 [15]. The expressed antibody fragments are presented as fusion proteins with the coat protein of M13 bacteriophage surfaces and allow selection against the targeted antigen. In contrast to traditional hybridoma technology, this in vitro selection method enables the creation of mAbs even against toxic and non-immunogenic agents. The impact of phage display on the development of therapeutic antibodies has been reviewed in detail by Frenzel et al. 2016 [16].
Figure 1.
Schematic overview of therapeutic mAb structure and its evolution from murine antibodies (dark blue domains) to fully human antibodies (light blue domains) with associated decrease in immunogenicity in humans. The constant domains of the heavy chain (CH2 and CH3) are localized in the Fc fragment. The Fab-fragments consist of the constant (CL) and variable (VL) parts of the light chains as well as the variable (VH) and constant domain (CH1) of the heavy chain. Adapted from [17].
Figure 1.
Schematic overview of therapeutic mAb structure and its evolution from murine antibodies (dark blue domains) to fully human antibodies (light blue domains) with associated decrease in immunogenicity in humans. The constant domains of the heavy chain (CH2 and CH3) are localized in the Fc fragment. The Fab-fragments consist of the constant (CL) and variable (VL) parts of the light chains as well as the variable (VH) and constant domain (CH1) of the heavy chain. Adapted from [17].
Further inventions in the field of structural modifications of mAbs were made by Lonberg et al. in 1994. By replacing the entire murine immunoglobulin repertoire in the mouse genome with those of humans it was possible to create fully humanized antibodies [18]. Such transgenic mice produce human-like antibodies after immunization followed by conventional hybridoma technology to obtain monoclonal antibodies. Panitumumab, the first fully human mAb isolated from a transgenic XenoMouse, was FDA-approved for therapeutic application in 2006 [19]. However, this approach is limited in cases where the immunogens are toxic or show a high degree of homology between targeted human antigen and its murine ortholog.
Despite of the mentioned limitations, creation of chimeric, humanized or fully human antibodies was a breakthrough and led to a wave of FDA-approved antibodies. Many of them belong to the best-selling pharmaceuticals worldwide, like the blockbuster Adalimumab with an effectiveness against rheumatoid arthritis and other chronic or immune-inflammatory diseases [20,21] (Table 1). Other relevant antibodies such as Pembrolizumab and Nivolumab are used in targeted melanoma therapy [22]. Among those common anti-cancer antibodies, modified antibody-drug conjugates (ADCs) were developed for targeting cancer-antigen and delivering small, chemically linked cytotoxic agents to tumor cells. Currently, only thirteen FDA-approved ADCs for numerous cancer variants were on the market [23]. Some major limitations during clinical trials were their heterogeneity due to non-specific drug conjugation and dynamic in vivo change after application [24]. Therefore, it is not surprising that many quantitative methods exist for the investigation of pharmacokinetic properties of ADCs in human plasma or serum [25,26,27]. With increasing market importance, analytical methods for the quantification of conventional therapeutic mAbs are also gaining relevance in clinical and pharmaceutical analysis.
2. Current Strategies in MS-Based Quantification of Antibodies
Over the last years, liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) became further established as one of the main technologies in antibody quantification [38]. Many innovative LC-MS/MS-based approaches were reported, focusing on more selective, more sensitive, faster and/or simplified detection of different antibody species in human blood. Most of the methods continued to quantify antibodies on a peptide level, however, studies directly measuring intact or partially digested antibodies were also increasingly presented. These methods are mainly used for the quantification of antibodies with potential matrix effects, for example in the clinical context of pharmacokinetic parameters such as antibody metabolism (half-life) or distribution in the patient’s body [39]. Moreover, several authors reported advancements in absolute amino acids analysis for the quantification of purified antibodies. In addition to the methods mentioned above, this strategy is particularly suitable for quality control of pharmaceutical antibody formulations in industry or accurate protein quantification of reference material products. Consequently, these reference materials can be employed to develop novel MS-based protein quantification techniques. Furthermore, they are metrologically traceable, thereby enhancing the comparability and reliability of measurement outcomes on a global scale.
An overview of the mentioned quantification strategies and target applications is provided in Figure 2. Developments in peptide-based methods will be discussed in detail in the next section; “intact” strategies and amino acid-based quantification will be shortly outlined after that.
2.1. Quantification of Enzymatically Digested Antibodies
A key advantage of peptide-based quantification is the high sensitivity and selectivity that can be achieved for these analytes on commonly available triple-quadrupole (QqQ) mass spectrometers. Typically, a complex biological sample such as human blood serum containing the target antibody (or antibodies) is enzymatically digested into numerous defined peptides of varying chain lengths. The mixture of peptides is separated chromatographically and one or more abundant, unique, antibody-specific peptides, also termed “signature peptides”, are analyzed by MS/MS. For quantification, peak intensities of signature peptides are related to those of matching standards, preferably represented by stable isotope-labeled (SIL) signature peptide, added to the sample in specific amounts. To date, no universally applicable protocol exists for peptide-based mAb quantification. Instead, individual method development and validation remain necessary for each mAb, as different sample characteristics affect digestion efficiency, peptide recovery, signal-to-noise ratios and so on[40]. This section reviews steps in sample preparation and peptide selection frequently reported as critical for robust quantification of mAbs in biological samples.
2.2.1. Purification and Enrichment
Due to interfering matrix components (proteins, lipids, DNA), LC-MS/MS analysis of antibodies in human serum usually requires an enrichment step in sample preparation. This can be carried out before or after the digestion step. Enrichment prior to digestion concentrates the mAb fraction and often uses magnetic beads or solid-phase extraction (SPE) cartridges, either coupled with anti-idiotypic antibodies for CDR-specific purification [41] or protein A and G that specifically bind to the constant Fc-region of mAbs [42]. Precipitation with organic solvents (e.g. methanol) or inorganic salts (ammonium sulfate) is an inexpensive and simple method for isolating the overall protein fraction prior to MS analysis [43]. Enrichment of targeted peptides after digestion is very commonly performed using C18-reversed phase/cation exchange SPE cartridges or tips, a step that also removes residual salts that may form excessive adducts during electrospray ionization (ESI).
It was also noted that each sample processing step carries the risk of analyte losses. For example, a significant sample loss of approx. 40% and poor quantitative precision of ± 15% was observed by Heudi et al. from SPE-based peptide cleanup of digested mAb [38]. This highlights the need to optimize SPE conditions individually for each assay and to include internal standards right from the beginning of the experiment.
2.2.2. Denaturation, Reduction, and Alkylation
Most published protocols include denaturation and reduction steps prior to digestion to unfold the tertiary structure and break intramolecular disulfide bonds of proteins. This facilitates access of the enzyme to the cleavage sites, ensuring efficient digestion. For the chemical denaturation of proteins, the use of buffers containing strong chaotropic denaturants such as urea [38] or guanidine hydrochloride [44,45] in a final concentrations >6 M and ionic detergents like sodium dodecyl sulfate (SDS) and sodium deoxycholate (DOC) [41,46] was reported. Proc et al. demonstrated that the use of guanidine hydrochloride consistently yielded lower digestion efficiency than urea, SDS or DOC-based denaturing protocols [47]. Additionally, guanidine hydrochloride can inhibit trypsin activity even at low concentrations, rendering a buffer exchange step necessary. High urea concentrations can also influence digestion efficiency, but sample dilution to a urea concentration <1 M was sufficient for complete trypsin digestion [38]. As urea is heat-sensitive, it can degrade in different buffers into ammonium cyanate, which binds to free amines by carbamylation reactions resulting in structure modifications of proteins [48]. However, if SDS remained in the buffer solution during trypsin digestion, the proteolytic enzyme denatured and subsequent MS analysis was affected by ionization suppression at levels as low as 0.01% [49,50].
Several MS-compatible detergents were made commercially available, such as ProteaseMax (Promega GmbH), RapiGest™ surfactant (Waters Corporation), PPS Silent Surfactant (Agilent Technologies) as well as Progenta (Protea Biosciences), which degrade proteins in combination with heat or in the low pH range [51]. For example, Abe et al. diluted a Nivolumab-containing sample with RapiGest™ surfactant and denatured by heating above 80°C [36]. Also, organic solvents were used at specific concentrations for enrichment by precipitation as well as sample denaturation without noticeable reduction in enzyme activity [47]. Each of the mentioned methods has its drawbacks and needs to be optimized experimentally.
The reduction step during denaturation is essential for allowing the proteolytic enzyme to access the entire protein structure. Dithiothreitol (DTT) is commonly used as a reduction agent [30,36,38,45,46]. As an alternative, tris(2-carboxyethyl)phosphine (TCEP) was used [52,53]. TCEP had higher pH as well as temperature stability and faster-reducing properties as compared to DTT. In the context of bottom-up sequencing, it was recently shown that the use of different reducing agents had only a minor impact on peptide identification [54]. By contrast, the influence of different alkylation agents on peptide yield was apparently larger. Alkylation agents derivatize free thiol groups of cysteines, thereby inhibiting their ability to reform disulfide bonds. The most frequently used alkylation agent for antibody quantification is iodoacetamide (IAM) [36,38,46,53]. The reaction is carried out in the absence of light and at room temperature to avoid side reactions. Over the past few years, different alkylation reactions were investigated in detail. Mueller and Winter compared IAM as an alkylation agent against iodoacetic acid (IAA), acrylamide (AA) and chloroacetamide (CAM) [55]. The authors identified unspecific reactions on the side chains of tyrosine, serine and threonine of digested HeLa proteins when using the iodine-containing reagents. Contrary to this, another study reported a higher yield of alkylated cysteine peptides and fewer side reactions when the procedure was performed using IAA compared to AA, N-ethylmaleimide or 4-vinylpyridine [54]. However, investigations by Kuznetsova et al. revealed that carbamidomethylation may affect up to 80% of peptides containing methionine after IAM-alkylation of digested proteins from HeLa and HepG2 cells [56]. Robinson and Hains also have found, that less light-sensitive chloroacetamide have an adverse impact on methionine oxidation [57]. Furthermore, IAM increased the rate of methionine-to-isothreonine conversion; structurally related IAA induced the same side reactions. Additionally, it has been demonstrated that IAM-alkylated peptides with N-terminal cysteines are prone to cyclization, which can result in a mass shift of -17 Da [58]. Consequently, optimization of reduction and alkylation conditions appears fundamental for antibody quantification. Due to the time-intensive optimization procedure as well as possible by-products, there are many protocols that skip these steps [28,29,30,35,37]. However, this can lead to incomplete protein digestion, as the antibody only partially unfolds or even refolds, and disulfide-connected peptides complicate the software-supported evaluation [59].
2.2.3. Digestion
Efficient enzymatic breakdown of therapeutic mAbs into peptides is crucial for reliable determination of the antibody amount by this approach. Specifically, digestion efficiencies appear to depend on the individual antibody structure, in particular with regard to phosphorylation and glycosylation patterns [59,60]. Proteases should be of the highest “MS grade” quality to ensure high specificity, activity, and purity. For MS analysis, peptides with basic residues at the C-terminus are preferred due to their enhanced ionization efficiency in positive electrospray ionization (ESI+) mode. Therefore, it is not surprising that the serine protease trypsin is the most frequently used proteolytic enzyme. Several trypsin products from different suppliers are available on the market (Table 2), varying in activity and optimal digestion conditions. It should be noted that naturally obtained trypsin from porcine or bovine pancreas can contain impurities of chymotrypsin. To prevent protease activity of chymotrypsin, the tosyl-phenylalanyl-chloromethyl-ketone (TPCK) is added to most commercial trypsin products. Furthermore, autolysis of trypsin results in the formation of pseudotrypsin with a chymotrypsin-like activity. Both drawbacks can be avoided by using recombinant and modified trypsin whose dimethylated lysine residues prevent self-digestion and increase stability [61,62]. Surface-immobilized trypsin is increasingly being implemented for antibody digestion, since it offers fast digestion with less self-digestion [36] and can be integrated as immobilized enzyme reactors for automated sample processing [63].
Trypsin cleaves proteins specifically at the C-terminus of the basic amino acids arginine (R) and lysine (K) (Figure 3), typically generating peptides of a size favorable for downstream LC–MS/MS analysis. Both amino acids are abundant and well-distributed in different proteins, producing peptides with an average length of 14 amino acids in length and a minimum of two positive charges [59]. However, when a proline residue follows at the C-terminus of R or K, the protein backbone is almost unaffected by trypsin. Furthermore, the presence of glutamic and aspartic acid as well as phosphorylated threonine and serine may increase the frequency of so-called missed cleavages. Therefore, some providers offer protease products combining standard trypsin with digestion of Lys-C (Table 2) which shares C-terminal K as cleavage site but continues to work under high urea concentrations (6–8 M urea). The combination minimizes missed K cleavages resulting in increased digestion efficiency and improved reproducibility [64].
Sometimes, trypsin fails to generate peptides of optimal length due to the high abundances of R and K in the antibody sequence. In such cases, the use of other proteases like chymotrypsin, AspN, GluC and ArgC can be good alternatives [65,66]. Here, the nomenclature of these enzymes corresponds to the target amino acid and terminus at which the protein is cleaved, respectively (Figure 3). Generally, enzymatic digestion should be performed in a buffer with compatible pH that is optimal for the specific enzyme. For example, the listed products in Table 2 recommend the use of ammonium bicarbonate or Tris buffers with pH between 7 to 8. However, Tris enhances the formation of various adduct ions and can cause ion suppression during ESI [50]. The selection of an appropriate buffer can also influence the generated peptides, whereby spontaneous chemical deamination of asparagine and glutamine residues may occur [67,68].
Furthermore, enzymatic proteolysis can be performed under different temperatures, digestion times and enzyme-to-antibody ratios. Typically, digestion with trypsin is carried out at 37 °C for one to 24 hours (Table 2). Some optimized or immobilized trypsin variants are stable at higher temperatures and enable faster digestion [59]. Additionally, the incubation time can depend on the position of target signature peptides and its steric accessibility. Especially peptides from CDRs located at the surface are easily accessible and allow fast enzymatic digestion [69]. Lastly, prolonged digestion time and elevated temperatures can introduce unwanted protein modifications and amino acid changes like asparagine deamidation and N-terminal glutamine cyclization [67,68,70] as well as oxidation [71]. For enzymatic digestion, an enzyme-to-antibody ratio of 1:100 to 1:20 is commonly recommended. Nevertheless, it is mandatory to check digestion specificity and completeness for absolute quantification.

Table 2.
Exemplary overview of commercially available MS-grade trypsin and trypsin/Lys-C variants, together with their recommended sample preparation procedures for LC-MS/MS analysis. Products from other manufacturers with similar properties may be equivalent. DTT: Dithiothreitol; TCEP: Tris(2-carboxyethyl)phosphine; IAA: Iodoacetic acid, IAM: Iodoacetamide; opt.: optional.
Table 2.
Exemplary overview of commercially available MS-grade trypsin and trypsin/Lys-C variants, together with their recommended sample preparation procedures for LC-MS/MS analysis. Products from other manufacturers with similar properties may be equivalent. DTT: Dithiothreitol; TCEP: Tris(2-carboxyethyl)phosphine; IAA: Iodoacetic acid, IAM: Iodoacetamide; opt.: optional.
| Product | Special Feature |
Denaturation | Reduction | Alkylation | Digestion conditions |
|---|---|---|---|---|---|
| Trypsin | |||||
|
Promega Trypsin Gold |
maximum specificity |
8 M Urea 1 hour |
DTT |
IAM 30 min |
Overnight 37°C |
| Rapid Digestion Trypsin | fast digestion | - | opt. | opt. | 1 h 70°C |
| Trypsin Platinum | recombinant enzyme, autoproteolytic resistance |
8 M GuHCl 30 min |
TCEP | IAM 30 min |
Overnight 37°C |
|
Thermo Fisher Pierce™ Trypsin |
|
1 hour at 60°C or 10 min at 95°C |
DTT |
IAA, 30 min |
4 to 24 h 37°C |
| SMART Digest Trypsin-Kit | automatable process | - | opt. | opt. | 45 min (IgG) 70°C |
| In-Solution Tryptic Digestion and Guanidination Kit | Improved ionization by guanidination of K into homo-R |
95°C 5 min |
DTT | IAM, 30 min |
2 hours at 37°C or overnight at 30°C |
|
Waters ProteinWorks eXpress Digest Kit |
High throughput of samples possible |
Digestion buffer, 80°C, 10 min |
Reduction Agent 60°C, 20 min |
Alkylation Agent 30 min |
2 h 45°C |
|
Promise Proteomics mAbXmise Kit |
Immunocapture cartridges |
opt., 4 M to 0.1 M Urea |
- |
- |
30 min to 15 h 37°C |
| Trypsin/ Lys-C Mix | |||||
|
Thermo Fisher EasyPep™ Mini MS Sample Prep Kit |
High throughput of samples possible |
Lysis Solution 95°C, 10 min |
Red. Solution |
Alk. Solution |
1 to 3 h 37°C |
| Pierce™ Trypsin/ Lys-C Protease Mix | 8 M Urea, 1 hour at 60°C or 10 min at 95°C |
DTT | IAM, 30 min |
2 to 16 hours 37°C |
|
|
Promega Rapid Digestion– Trypsin/LysC |
Fast digestion |
- |
opt. |
opt. |
1 hour 70°C |
| Trypsin/Lys-C | Quantification | 6-8 M Urea, 30 min |
DTT | IAM, 30 min |
overnight 37°C |
2.2.4. Signature Peptide Selection
Quantification of mAbs on a peptide level requires the selection of suitable signature peptides that allow both selective and sensitive detection of the target antibody in the sample matrix (Table 1). Knowledge of the amino acid sequence of the target antibody is advantageous for this selection. Generally, protein sequences of FDA-approved mAbs are available in the Immunogenetics Information system® (http://www.imgt.org/) or databases of ABCD (AntiBodies Chemically Defined, https://web.expasy.org/abcd/). For prediction of potentially generated peptides, the free programs MS-DIGEST from Protein Prospector (http://prospector.ucsf.edu) or DeepDigest [72] can be used. When quantifying antibodies in human serum, peptides of the CDR are preferable due to their uniqueness for each mAb as well as enzymatic accessibility. Accordingly, predicted peptide sequences can be analyzed with Basic Local Alignment Search Tool for proteins (https://blast.ncbi.nlm.nih.gov/Blast.cgi) or Peptide Atlas (http://www.peptideatlas.org) for matches with interfering peptide sequences of the human proteome. For quantification of purified therapeutic antibodies, choosing of peptides from the constant Fc-region can also be used, allowing the quantification of multiple mAbs of similar subclass with the same MS assay.
Nevertheless, the selection of suitable signature peptides for quantitative measurements can be affected by some analytical and equipment limitations. Initially, the selected peptides should have a length between eight to 25 amino acids. Often, shorter sequences are not unique, while longer sequences usually lead to a significant charge distribution [66]. In addition, the amino acids contained in the peptide candidates should be considered based on their susceptibility of chemical modifications and instability that has become known in recent years [73,74]. For example, cysteine, methionine, tryptophan, and histidine are well-known amino acids prone to oxidation and leading to mass shift of +15.9949 Da. Furthermore, asparagine or glutamine can undergo deamidation during trypsin digestion, causing mass shifts of -17.0265 Da and +0.9840 Da, respectively. The loss of ammonia through N-terminal glutamine cyclization induced by prolonged digestion time also leads to loss of -17.0265 Da. It should also be noted, that therapeutic antibodies can undergo oxidation or deamination during production, purification or storage and may therefore already be present before enzymatic digestion [73]. In this context, signature peptides that are rich in potentially modifiable amino acids should be avoided. Other important tips for selecting the right signal peptides for targeted protein quantification have been discussed elsewhere [75,76,77].
Likewise, peptides potentially carrying glycan residues should be treated with care due their influence of trypsin cleavage efficiency [60]. Software tools such as NetNglyc [78](https://services.healthtech.dtu.dk/services/NetNGlyc-1.0/), NGlyc [79] (https://github.com/bioinformaticsML/Ngly) or newer deep neural network-based approach DeepNGlyPred [80] (https://github.com/dukkakc/DeepNGlyPred) can be used to check peptide sequences for potential glycosylation issues.
2.2. Quantification of Intact Antibodies
In contrast to peptide-based quantification, this strategy enables the direct quantification of intact antibodies without digestion. This approach has proven particularly useful for monitoring the biotransformation of therapeutic mAbs in patients. Jian et al. published a method for absolute quantification of intact mAbs in plasma, using an isotope-labeled mAb as internal standard and automated software-assisted mass peak deconvolution [81]. Selective preconcentration and sample clean-up of targeted antibodies from interfering matrix was achieved by immunoaffinity capture (IAC). Similarly, a ligand binding assay (LBA) was integrated into a High-resolution(HR)MS workflow for simultaneous quantification of different human IgG1s at intact level, including a (13C)-labeled variant as internal standard [82]. The authors validated the method regarding selectivity, sensitivity, accuracy and precision, carryover, dilution linearity as well as reproducibility and found similar performance compared to peptide-based quantification. In addition, deglycosylation of intact human IgG1 with the enzyme PNGase F resulted in a less complex full-scan MS spectrum and increased the signal for each charge state. Notably, the use of PNGase F also allows the parallel quantification of N-glycan structures, which are involved in many biological processes. Methodological concepts for this purpose have been discussed elsewhere [83,84].
However, absolute quantification of intact mAbs by LC-MS/MS involves a range of drawbacks. The main problems are related to the high molecular weight [85] and molecular heterogeneity due to post-translational and non-enzymatic modifications [86]. As the complexity of charge state and isotope distributions increases with mass in ESI mass spectra, signal intensity decreases, resulting in a loss of analytical sensitivity. Although deconvoluted spectra can circumvent this problem to some degree [87,88], their use for quantification purposes is currently not recommended due to potential issues in data processing [85]. Furthermore, the necessary enrichment of mAbs with IAC prior to LC-MS/MS analysis adds additional complexity and has been critically reviewed by Zhao et al. [89]. If IAC enrichment is unavoidable and SIL internal standard is used for quantification, its addition to the matrix prior to sample preparation is compulsory [90]. For quantification of intact mAbs, the commercially available isotope labeled human IgG SILu™ mAb has been applied in a few studies [81,91]. Furthermore, the mAbXmise kit from Promise Proteomics (Table 2) also contains SIL-mAbs for frequently occurring antibodies in the field of inflammatory and oncological therapies, e.g. Adalimumab [32,92]. Similarly, in-house production of SIL analogues of targeted mAbs was also investigated [82].
2.3. Quantification of Hydrolysed Antibodies
Amino acid analysis (AAA) refers to a broadly used, absolute quantification method for targeting amino acids hydrolytically released from proteins and peptide samples. Commonly, hydrolysis of 1 to 10 nmol of protein is carried out under acidic as well as oxygen-free conditions using 6 M hydrochloric acid (HCl), elevated temperatures between 90 to 150 °C and incubation times ranging from 1 to 72 h [93,94,95]. To reduce reaction time, application of microwave assisted heating is widely used [95]. However, substances like salts, metal ions or urea in the hydrolysis solution can potentially affect the release of certain amino acids and should therefore be avoided [93]. Phenol, by contrast, can prevent amino acid degradation and halogenation of tyrosine during the hydrolysis process [93,95].
The preferred LC modes used for AAA of hydrolyzed proteins are hydrophilic interaction chromatography (HILIC), ion exchange (IEX) or reversed phase (RP) chromatography [96]. Conventionally, AAA included a derivatization step to optimize chromatographic separation as well as UV sensitivity for UV/VIS detection. With advances in LC-MS/MS analysis, accurate quantification of underivatized amino acids has now become widespread. The omission of derivatization is beneficial, as it eliminates the risk of potential contamination that may arise during the derivatization process [97].
Analogous to absolute quantification of peptides, LC-MS/MS allows absolute AAA by including SIL internal standards in the analysis. Here, SIL internal standards can be amino acids [98,99], peptides or full-length protein analogues [100]. When certified reference materials (CRMs) are used, measurements become fully traceable to Système International d’Unités (SI units) [95]. By using four SIL amino acids as internal standards in an LC-MS/MS setup, Jeong et al. developed an accurate quantification method for human growth hormone with an intra- and inter-day precision (RSD) of less than 1% [101]. Accordingly, the absolute quantification of proteins by AAA allows traceable and comparable results, as well as the certification of therapeutic protein products with certified SIL reference materials. Interlaboratory comparison for therapeutic antibody quantification [102] have proven the validity of this technique. Furthermore, this approach has also contributed to the development of several protein reference materials, such as COVID-19 analytical reagents [98,99,103].
3. Selection of Internal Standards for Quantification with LC-MS/MS
The choice of suitable internal standards is critical for quantitative MS, as losses during digestion and hydrolysis as well as matrix effects can lead to significant technical bias in quantification. SIL internal standards contain at least one stable isotope (usually 13C, 15N or D) in at least one amino acid which usually ensures co-elution with the respective target peptide or amino acid. Accurate quantification of mAbs was demonstrated using both SIL proteins and peptides, which in turn can be quantified by means of SIL peptides or amino acid standards. To enable metrological comparability and reliability of measurement results, calibration should be performed using SIL internal standards that can be traced back to reference materials.
3.1. Intact Antibody Standards
Different SIL antibody standards were presented for mAb quantification at the intact level. Mainly, commercially available SIL-mAbs were used [81,91] and offered from different suppliers like Promise Proteomics [104] or Sigma Aldrich. The use of full-length [32,37] or partial [28,33] SIL analogues for digested mAbs were also tested and included in quantification protocols. These internal standards can be calibrated using reference materials of known quantity, the so-called PSAQ method [100,105]. For example, Lebert et al. demonstrated simultaneous quantification of several therapeutically relevant and structurally similar IgG1 and IgG4 isotypes using PSAQ [52]. For in-house production of SIL mAbs, the antibody-producing cell line is allowed to grow in media containing isotopologues of one or more amino acids, which are incorporated into the desired mAb [106]. Although SIL amino acids are readily available, the approach is usually cost- and time-intensive [90].
A more cost-effective alternative for peptide-based quantification with antibody standards is to use SIL mAbs that produce identical or similar peptides in enzymatic digestion, allowing multiple therapeutic antibodies to be quantified simultaneously. For instance, the quantification of four therapeutic antibodies using SIL mAb analogues showed sufficient precision (CV) of less than 20% when signature peptides had similar amino acid sequence and were located in the VH-region [107]. Furthermore, the purchasable human IgG1 SILuMAb with SIL arginine (R) and lysine (K) residues can also be a less expensive alternative for peptide-based quantification due a common FC region with other therapeutic mAbs. However, as noted by Smit and co-workers, the digestion kinetics can vary between SIL mAbs and the mAbs of interest when different recombinant expression systems were used, likely due to associated variations in post-translational modifications [108].
In summary, internal standardization with SIL mAbs can provide crucial advantages in terms of reproducibility and robustness. Due to the identical behavior of targeted and SIL antibody, throughout the entire analytical workflow, variability and inferences in MS analysis can be reduced to a minimum. However, a significant drawback is the overall cost and quality of SIL antibody standards, which currently restricts their broader application.
3.2. Peptide Standards
SIL peptide standards generally share the same AA sequence with the selected signature peptides. SIL peptides can be easily synthesized in HPLC-grade purity and are thus much cheaper than intact SIL mAbs. SIL peptide-based mAb quantification has become common practice over the last decade [36,42,46,53]. An important factor is the concentration and purity of SIL peptides that should be verified by AAA to obtain precise reference values [95,109]. Burkitt et al. established a method for protein quantification in which the SIL peptide standards were quantified by isotope dilution MS (IDMS) [110]. By using certified AA reference materials, mAb quantification was fully traceable to SI units.
In contrast to antibody standards, SIL peptides can only correct for variability in instrumental factors and matrix effects but not for changes in sample purification or in enrichment prior to digestion. Extended SIL peptides contain additional amino acids adjacent to the cleavage site of the signature peptide. They can improve tracing digestion efficiency as additional amino acids need to be eliminated during enzymatic cleavage. Benesova at el. demonstrated that SIL peptides extended by three amino acids at each N- and C-terminus, enable quantification results for albumin equivalent to those based on a SIL protein [111]. However, digestion kinetics can also differ for targeted mAbs and extended SIL peptides due to accessibility of the cleavage site. For example, Li et al. obtained higher precision with a SIL intact antibody (< 16% CV) than with SIL peptides in both standard and extended variants (> 25%) [112]. Therefore, it is often recommended to measure at least two signature peptides to compare cleavage rate and to produce more robust results [42,113].
Peptide modifications occurring during sample preparation are another factor that can prevent accurate quantification of mAbs. For instance, CDR-specific SIL peptide standards prone to methionine oxidation impaired the quantification of therapeutic mAbs [71]. Similarly, the quantification of growth hormone indicated that the addition of the SIL peptide prior to digestion was crucial for correcting peptide degradation rates [113].
3.3. Amino Acid Standards
As previously described [93], only a subset of the 20 proteinogenic amino acids have to be proven to be sterically accessible and generally stable enough for the acidic hydrolysis procedure. For example, peptide bonds between aliphatic/hydrophobic amino acids are difficult to break, resulting in insufficient release and underestimation without additional hydrolysis time. Furthermore, the chemical oxidation of methionine or deamination of asparagine and glutamine into their acid counterparts make these amino acids unsuitable for quantification. Although correction factors were developed, the extent of hydrolysis losses and degradation artefacts were shown to vary between laboratories. Therefore, absolute protein quantification by AAA is currently limited to specific amino acids, with proline, valine, isoleucine, leucine, and phenylalanine being favored in the literature [98,99,102,103,110].
4. Software Tools Supporting Targeted mAb Quantification
The commonly used acquisition mode for absolute mAb quantification at the peptide or amino acid level is multiple reaction monitoring (MRM), using highly sensitive and selective QqQ or quadrupole-ion trap (Q-Trap) mass spectrometers. Although targeted peptides or amino acids and their spiked SIL analogues exhibit nearly identical physicochemical properties (e.g., retention time, fragmentation patterns), the MS1 monoisotopic mass shifts depending on the incorporation of labeled isotopes. Quantification based on MRM involves extracted ion chromatograms (XICs) that are created for both the precursor and product m/z of the selected peptides or amino acids. By relating peak areas of added SIL standard to those of the target peptides or amino acids, the antibody concentration can be precisely determined. The development of efficient MRM schemes depends on many critical factors such as chromatographic separation, selectivity of transitions as well as fine-tuned fragmentation energies. Furthermore, metrological parameters including limit of detection and quantification (LOD, LOQ), accuracy, precision, linearity and stability, following FDA [116] or EMA [117] guidelines, must be taken into consideration during method development. Necessary procedures for the latter have already been discussed in detail for beginners elsewhere [76,118]. Here, we focus on software packages that support method development and data analysis but can themselves pose a challenge for inexperienced users in the field. Software tools enabling time-efficient and user-friendly processing of large sample numbers, obtained from different LC-MS/MS setups, are also discussed. Furthermore, solutions for the automated prediction of tryptic peptides and their organization into spectral libraries are addressed.
4.1. Commercial and Device-Specific Software
All leading MS manufacturers offer workflow-oriented software solutions supporting mAb characterization, partially in combination with pre-configured instrument stacks, such as the BioAccord System (Waters). The latter is specifically designed for routine and automated monitoring of biotherapeutics in quality control processes, including multi-attribute method (MAM) workflows for analyzing antibody degradation and modification e.g. during storage [119] and quantification of mAbs at the intact level [120]. More general software for MRM analysis by the same manufacturer includes the TargetLynx™ Application Manager (method development, automated sample data acquisition, processing, and reporting) and QuanOptimize™ (automated optimization of MRM transitions and collision energies). Similarly, BioPharma FinderTM (ThermoFisher) is a comprehensive software package offering pre-defined workflows, e.g. enabling quantification at the intact level due to meticulous deconvolution of mass spectra or multi-attribute characterization of mAbs in contexts such as bioprocessing up to final product quality control [35,121,122]. To identify proteotypic peptides and predict fragmentation patterns for targeted quantification, PinpointTM (ThermoFisher) helps to determine MRM transitions and can export methods directly to compatible instruments. Due to an iterative workflow design, acquired data can be used to refine acquisition methods and verify top peptide candidates [123,124]. MassHunter (Agilent) includes various packages for data acquisition and quantitative analysis. MassHunter Optimizer supports the development of MRM methods by automatically optimizing acquisition parameters, facilitating e.g. the development of more sensitive triggered MRM (tMRM) methods [125]. The Analyst® software suite (Sciex) not only provides general MS data acquisition and analysis but also offers a variety of tools specifically designed for the analysis of mAbs. These tools include intact mass spectrum characterization, subunit analysis, peptide mapping, glycan analysis, and DAR calculations (Biologics Explorer, ProteinPilotTM). In addition, MRMPilotTM supports building and optimizing peptide MRM experiments, allowing inspection of full scan MS/MS data as well as transferring transition settings to the acquisition module. MRMPilotTM also allows input from open databases such as PeptideAtlas [126]. Similarly to MassHunter, it also supports the developing of MRM-triggered acquisition methods. For data evaluation, MultiQuantTM (Sciex) supports absolute quantification based on multiple peptides and the creation of reports including common quality parameters such as the coefficient of variation (CV) and accuracy.
Furthermore, third-party software primarily aimed at high-throughput proteomics can be applied to targeted quantification tasks. Available options include SpectronautTM, SpectromineTM and SpectroDiveTM (Biognosis) as well as Mascot Distiller (Matrix Science). These software packages accept data from all major MS instruments.
4.2. Open-Source Software Alternatives
In addition to commercially available software solutions, several open-source programs and web-based applications have become available for MRM method development and data analysis. One of the most popular options covering the entire experimental workflow is Skyline, which was released by the MacCoss group in 2009 [127]. The software is manufacturer-independent and is designed for the quantification of small molecules, including amino acids and peptides. The software enables the comparison of MS/MS spectra with integrated databases and the development of targeted MRM methods. The functionality of the software is subject to continuous improvement based on user feedback. It includes the prediction of peptide ions and their transitions based on the uploaded antibody sequence, selected modifications (isotopic labels, PTMs), digestion enzyme and instrument-specific requirements. Furthermore, the software enables the prediction of retention times and facilitates the construction of spectral libraries with Prosit [128]. A range of free tutorials provide entry points for new users to the various aspects of data analysis.
Another freely available and widely used alternative for data evaluation is MaxQuant, developed by groups around Jürgen Cox and Matthias Mann at the Max Planck Institute of Biochemistry [129]. Analogous to Skyline, it is designed to analyze high-throughput mass-spectrometry data in a desktop application It supports various labeling techniques and algorithms for peak detection, as well as peptide identification with corresponding MS2 identification (Andromeda). However, it is more suitable for high-resolution mass spectrometry (HRMS) data than for multiple reaction monitoring (MRM) data. A substantial collection of published protocols is available for the implementation of MaxQuant in one’s own pipeline for the analysis of mass spectrometric data [130,131].
In addition to the previously discussed options, there are other, more sophisticated software solutions that will be briefly outlined here. OpenMS differs from Skyline in that it provides a flexible programming framework (Python) rather than a ready-to-use desktop interface [132,133]. Functionality includes core algorithms for MS data analysis such as peak detection, alignment, integration etc. as well as predefined high-level workflows. MRMPROBS, again, is a stand-alone desktop application aimed at automatic detection and identification of MRM signals based on probabilistic criteria [134]. Features include XIC panels for efficient quality control (QC) and manual peak correction, but no direct support for peptide targets is provided; target lists must be provided as a text file. MassChroQ is a command-line program supporting both low resolution (MRM) and HRMS data, as peak quantification is based on XICs rather than feature detection [135]. Peaks from all samples are automatically aligned and exported to a spreadsheet format, allowing integration into data pipelines. MRMAnalyzer similarly focusses on automated pipelines but is implemented as an R package [136,137]. Additionally, MassIVE.quant [138], quantms [139] and MRMPro [140] are examples of recent cloud-based platforms for quantitative proteomics, featuring large-scale online data processing capabilities and extensive QC plots for peak inspection.
5. Outlook: Need for Standardized Protocols, Certified Reference Materials, and New Technologies
The use of recombinant antibodies in both therapeutic and diagnostic applications will continue to increase over the next few years. Their exact quantification, both in pure form and in serum, plasma, or blood samples, is therefore essential. As shown in the previous chapters, a variety of peptide-based methods and protocols have been developed in recent decades with different digestion conditions or SIL materials and for each commercial antibody individually (Table 1). The need for standardized methods was impressively demonstrated in a study about the quantification of a recombinant human IgG1 SARS-CoV-2 antibody by the Protein Analysis Working Group (PAWG) of the Comité Consultatif pour la Quantité de Matière (CCQM) [102,141]. Participating national metrology institutes (NMIs) showed that there was a large discrepancy in the results (up to 22% difference from reference value) despite the use of ID-MS for peptide-based quantification approaches. Based on this, the digestion conditions and peptide selection for this SARS-CoV-2 antibody were optimized and validated with a reference antibody and AAA as reference method [53]. The authors clearly demonstrated how important it is to use CRMs to develop robust methods and generate accurate results. The need for suitable reference materials and standardized measurements is also emphasized in the clinical field for the quantification of biomarkers, e.g. apolipoprotein A [142,143,144]. The implementation of reference materials for quantitative LC-MS/MS assay development enables metrological traceability of calibration standards, ensuring that measurement results have a documented and unbroken chain of traceability to SI units [108]. Furthermore, it can also compensate for differences in the device-specific ionizations or software-dependent peak detection and smoothing settings.
A broadly accessible metrological reference material would provide a representative standard to enhance the harmonization of antibody quantification measurements [145] . For the development of standardized methods, the two certified antibody reference materials NISTmAb (RM 8671) and AISTmAb (RM 6208-a) from the National Institute of Standards and Technology (NIST, USA) and National Institute of Advanced Industrial Science and Technology (AIST, Japan) are available [146,147]. Due to the extensive effort needed to establish “certified” values, only a limited number of analytes are certified as reference material. The necessity for CRMs has also become especially evident during COVID-19 pandemic, particularly in the context of validating analytical tests [98,99,103]. Besides, the WHO also established several antibody reference standards, including the WHO adalimumab reference standard (ref. 17/236) and the WHO infliximab reference standard (Re 16/170). However, these standards exhibit inaccuracies in their concentrations [32], highlighting the need for certified, traceable materials which should be characterized through multidisciplinary approaches and evaluated by various laboratories [148]. When selecting suitable calibrators, it is essential to consider the differences in PTMs, as these variations can lead to potential deviations in measurement results [149].
Apart from method standardization and CRM development, new technologies may also contribute to faster and more streamlined assay development. We regard innovations in two fields as particularly promising. The first field relates to innovative enzyme technologies. A recent method that employs aqueous microdroplet-mediated enzymatic digestion has shown a remarkable decrease in digestion times, achieving results in the millisecond range, in contrast to the several hours typically required by conventional method [150]. Moreover, this approach has achieved a very high sequence coverage. Similarly, recombinant and immobilized enzyme systems have demonstrated potential in boosting cleavage rates, apparently due to improved stability, inhibition of trypsin self-digestion and higher enzyme-to-protein ratios [59,62]. Furthermore, there are also initial publications on thermostable enzyme variants, for example by inserting mutations [151] or glycosylation [152]. We believe that significant further optimization of enzyme systems is feasible due to progress in biotechnology, material science and AI-supported designs. Thereby, the dependency of digestion efficiency on the primary sequence and PTMs of mAb targets could be minimized, resulting in much improved commutability of calibrator and SIL materials.
A second field where innovation can have a significant impact on future mAb assays pertains to the production of SIL mAbs. The recently announced mAbXmise kit [92] offers a convenient mixture of several SIL mAbs, enabling the simultaneous quantification of various therapeutic mAbs [32]. The development of further mAb kits may result in more efficient production pipelines, thereby enhancing the broader and more affordable availability of SIL mAbs. This approach could improve compensation for parameters such as digestion conditions and purification from complex matrices, leading to more reproducible results. Ideally, an increase in demand could lead to the commercial availability of SIL mAbs becoming comparable to that of SIL peptides. However, production is expected to remain more complex and costly in the foreseeable future. Cell-free in vitro systems, such as PURE (Protein synthesis Using Recombinant Elements), could provide a cost-effective solution for the production of SIL mAbs [153].
Author Contributions
Conceptualization and review writing, S.D.; conceptualization, writing and supervision, Z.K. and C.J.; critical reading and editing, M.G.W and Y.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Special thanks to Gaby Bosc-Bierne for critical reading and remarks.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495-497. [CrossRef]
- Todd, P.A.; Brogden, R.N. Muromonab CD3. A review of its pharmacology and therapeutic potential. Drugs 1989, 37, 871-899. [CrossRef]
- Schroff, R.W.; Foon, K.A.; Beatty, S.M.; Oldham, R.K.; Morgan Jr, A.C. Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer research 1985, 45, 879-885.
- Jaffers, G.J.; Fuller, T.; Cosimi, A.; Russell, P.; Winn, H.; Colvin, R. Monoclonal antibody therapy. Anti-idiotypic and non-anti-idiotypic antibodies to OKT3 arising despite intense immunosuppression. Transplantation 1986, 41, 572-578. [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proceedings of the National Academy of Sciences 1984, 81, 6851-6855. [CrossRef]
- Brüggemann, M.; Winter, G.; Waldmann, H.; Neuberger, M.S. The immunogenicity of chimeric antibodies. Journal of Experimental Medicine 1989, 170, 2153-2157. [CrossRef]
- Faulds, D.; Sorkin, E.M. Abciximab (c7E3 Fab). A review of its pharmacology and therapeutic potential in ischaemic heart disease. Drugs 1994, 48, 583-598. [CrossRef]
- Leget, G.A.; Czuczman, M.S. Use of rituximab, the new FDA-approved antibody. Current opinion in oncology 1998, 10, 548-551. [CrossRef]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522-525. [CrossRef]
- Queen, C.; Schneider, W.P.; Selick, H.E.; Payne, P.W.; Landolfi, N.F.; Duncan, J.F.; Avdalovic, N.M.; Levitt, M.; Junghans, R.P.; Waldmann, T.A. A humanized antibody that binds to the interleukin 2 receptor. Proceedings of the National Academy of Sciences 1989, 86, 10029-10033. [CrossRef]
- Wiseman, L.R.; Faulds, D. Daclizumab: a review of its use in the prevention of acute rejection in renal transplant recipients. Drugs 1999, 58, 1029-1042. [CrossRef]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods – a review and update. Biotechnology and Genetic Engineering Reviews 2013, 29, 175-186. [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: filamentous phage displaying antibody variable domains. Nature 1990, 348, 552-554. [CrossRef]
- Smith, G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315-1317. [CrossRef]
- Zhang, Y. Evolution of phage display libraries for therapeutic antibody discovery. In Proceedings of the MAbs, 2023; p. 2213793.
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. In Proceedings of the MAbs, 2016; pp. 1177-1194.
- Sánchez-Robles, E.M.; Girón, R.; Paniagua, N.; Rodríguez-Rivera, C.; Pascual, D.; Goicoechea, C. Monoclonal Antibodies for Chronic Pain Treatment: Present and Future. International Journal of Molecular Sciences 2021, 22, 10325. [CrossRef]
- Lonberg, N.; Taylor, L.D.; Harding, F.A.; Trounstine, M.; Higgins, K.M.; Schramm, S.R.; Kuo, C.-C.; Mashayekh, R.; Wymore, K.; McCabe, J.G.; et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature 1994, 368, 856-859. [CrossRef]
- Jakobovits, A.; Amado, R.G.; Yang, X.; Roskos, L.; Schwab, G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nature Biotechnology 2007, 25, 1134-1143. [CrossRef]
- Lapadula, G.; Marchesoni, A.; Armuzzi, A.; Blandizzi, C.; Caporali, R.; Chimenti, S.; Cimaz, R.; Cimino, L.; Gionchetti, P.; Girolomoni, G.; et al. Adalimumab in the Treatment of Immune-Mediated Diseases. International Journal of Immunopathology and Pharmacology 2014, 27, 33-48. [CrossRef]
- Verdin, P. Top companies and drugs by sales in 2023. Nature reviews. Drug Discovery 2024. [CrossRef]
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.-O. Current Status and Future Directions of the Immune Checkpoint Inhibitors Ipilimumab, Pembrolizumab, and Nivolumab in Oncology. Annals of Pharmacotherapy 2015, 49, 907-937. [CrossRef]
- Liu, K.; Li, M.; Li, Y.; Li, Y.; Chen, Z.; Tang, Y.; Yang, M.; Deng, G.; Liu, H. A review of the clinical efficacy of FDA-approved antibody‒drug conjugates in human cancers. Molecular Cancer 2024, 23, 62. [CrossRef]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. mAbs 2014, 6, 34-45. [CrossRef]
- Zhu, X.; Huo, S.; Xue, C.; An, B.; Qu, J. Current LC-MS-based strategies for characterization and quantification of antibody-drug conjugates. Journal of Pharmaceutical Analysis 2020, 10, 209-220. [CrossRef]
- Todoroki, K.; Yamada, T.; Mizuno, H.; TOYO’OKA, T. Current mass spectrometric tools for the bioanalyses of therapeutic monoclonal antibodies and antibody-drug conjugates. Analytical Sciences 2018, 34, 397-406. [CrossRef]
- Cahuzac, H.; Devel, L. Analytical Methods for the Detection and Quantification of ADCs in Biological Matrices. Pharmaceuticals 2020, 13, 462. [CrossRef]
- Millet, A.; Khoudour, N.; Guitton, J.; Lebert, D.; Goldwasser, F.; Blanchet, B.; Machon, C. Analysis of Pembrolizumab in Human Plasma by LC-MS/HRMS. Method Validation and Comparison with Elisa. Biomedicines 2021, 9, 621. [CrossRef]
- Hallin, E.I.; Serkland, T.T.; Bjånes, T.K.; Skrede, S. High-throughput, low-cost quantification of 11 therapeutic antibodies using caprylic acid precipitation and LC-MS/MS. Analytica Chimica Acta 2024, 342789. [CrossRef]
- de Jong, K.A.; Rosing, H.; Huitema, A.D.; Beijnen, J.H. Optimized sample pre-treatment procedure for the simultaneous UPLC-MS/MS quantification of ipilimumab, nivolumab, and pembrolizumab in human serum. Journal of Chromatography B 2022, 1196, 123215. [CrossRef]
- Willeman, T.; Jourdil, J.-F.; Gautier-Veyret, E.; Bonaz, B.; Stanke-Labesque, F. A multiplex liquid chromatography tandem mass spectrometry method for the quantification of seven therapeutic monoclonal antibodies: application for adalimumab therapeutic drug monitoring in patients with Crohn’s disease. Analytica chimica acta 2019, 1067, 63-70. [CrossRef]
- Tron, C.; Lemaitre, F.; Bros, P.; Goulvestre, C.; Franck, B.; Mouton, N.; Bagnos, S.; Coriat, R.; Khoudour, N.; Lebert, D.; et al. Quantification of infliximab and adalimumab in human plasma by a liquid chromatography tandem mass spectrometry kit and comparison with two ELISA methods. Bioanalysis 2022. [CrossRef]
- Scheffe, N.; Schreiner, R.; Thomann, A.; Findeisen, P. Development of a mass spectrometry-based method for quantification of ustekinumab in serum specimens. Therapeutic Drug Monitoring 2020, 42, 572-577. [CrossRef]
- Yamaoka, K.; Irie, K.; Hiramoto, N.; Hirabatake, M.; Ikesue, H.; Hashida, T.; Shimizu, T.; Ishikawa, T.; Muroi, N. Safety and blood levels of daratumumab after switching from intravenous to subcutaneous administration in patients with multiple myeloma. Investigational New Drugs 2023, 41, 761-767. [CrossRef]
- Li, W.; Huang, W.; Yu, X.; Chen, C.; Yuan, Y.; Liu, D.; Wang, F.; Yu, J.; Diao, X. A validated LC-MS/MS method for the quantitation of daratumumab in rat serum using rapid tryptic digestion without IgG purification and reduction. Journal of Pharmaceutical and Biomedical Analysis 2024, 116083. [CrossRef]
- Abe, K.; Shibata, K.; Naito, T.; Karayama, M.; Hamada, E.; Maekawa, M.; Yamada, Y.; Suda, T.; Kawakami, J. Quantitative LC-MS/MS method for nivolumab in human serum using IgG purification and immobilized tryptic digestion. Analytical Methods 2020, 12, 54-62. [CrossRef]
- Millet, A.; Khoudour, N.; Bros, P.; Lebert, D.; Picard, G.; Machon, C.; Goldwasser, F.; Blanchet, B.; Guitton, J. Quantification of nivolumab in human plasma by LC-MS/HRMS and LC-MS/MS, comparison with ELISA. Talanta 2021, 224, 121889. [CrossRef]
- Heudi, O.; Barteau, S.; Zimmer, D.; Schmidt, J.; Bill, K.; Lehmann, N.; Bauer, C.; Kretz, O. Towards Absolute Quantification of Therapeutic Monoclonal Antibody in Serum by LC−MS/MS Using Isotope-Labeled Antibody Standard and Protein Cleavage Isotope Dilution Mass Spectrometry. Analytical Chemistry 2008, 80, 4200-4207. [CrossRef]
- Wei, C.; Su, D.; Wang, J.; Jian, W.; Zhang, D. LC–MS challenges in characterizing and quantifying monoclonal antibodies (mAb) and antibody-drug conjugates (ADC) in biological samples. Current Pharmacology Reports 2018, 4, 45-63. [CrossRef]
- Hentschel, A.; Piontek, G.; Dahlmann, R.; Findeisen, P.; Sakson, R.; Carbow, P.; Renné, T.; Reinders, Y.; Sickmann, A. Highly sensitive therapeutic drug monitoring of infliximab in serum by targeted mass spectrometry in comparison to ELISA data. Clinical Proteomics 2024, 21, 16. [CrossRef]
- Xu, K.; Liu, L.; Maia, M.; Li, J.; Lowe, J.; Song, A.; Kaur, S. A multiplexed hybrid LC–MS/MS pharmacokinetic assay to measure two co-administered monoclonal antibodies in a clinical study. Bioanalysis 2014, 6, 1781-1794. [CrossRef]
- Chiu, H.-H.; Liao, H.-W.; Shao, Y.-Y.; Lu, Y.-S.; Lin, C.-H.; Tsai, I.-L.; Kuo, C.-H. Development of a general method for quantifying IgG-based therapeutic monoclonal antibodies in human plasma using protein G purification coupled with a two internal standard calibration strategy using LC-MS/MS. Analytica chimica acta 2018, 1019, 93-102. [CrossRef]
- Ouyang, Z.; Furlong, M.T.; Wu, S.; Sleczka, B.; Tamura, J.; Wang, H.; Suchard, S.; Suri, A.; Olah, T.; Tymiak, A. Pellet digestion: a simple and efficient sample preparation technique for LC–MS/MS quantification of large therapeutic proteins in plasma. Bioanalysis 2012, 4, 17-28. [CrossRef]
- Yang, Z.; Hayes, M.; Fang, X.; Daley, M.P.; Ettenberg, S.; Tse, F.L. LC− MS/MS Approach for Quantification of Therapeutic Proteins in Plasma Using a Protein Internal Standard and 2D-Solid-Phase Extraction Cleanup. Analytical chemistry 2007, 79, 9294-9301. [CrossRef]
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Bones, J. Multi-attribute method (MAM) to assess analytical comparability of adalimumab biosimilars. Journal of Pharmaceutical and Biomedical Analysis 2023, 234, 115543. [CrossRef]
- El Amrani, M.; Gerencser, L.; Huitema, A.D.; Hack, C.E.; van Luin, M.; van der Elst, K.C. A generic sample preparation method for the multiplex analysis of seven therapeutic monoclonal antibodies in human plasma or serum with liquid chromatography-tandem mass spectrometry. Journal of Chromatography A 2021, 1655, 462489. [CrossRef]
- Proc, J.L.; Kuzyk, M.A.; Hardie, D.B.; Yang, J.; Smith, D.S.; Jackson, A.M.; Parker, C.E.; Borchers, C.H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. Journal of proteome research 2010, 9, 5422-5437. [CrossRef]
- Sun, S.; Zhou, J.-Y.; Yang, W.; Zhang, H. Inhibition of protein carbamylation in urea solution using ammonium-containing buffers. Analytical Biochemistry 2014, 446, 76-81. [CrossRef]
- Loo, R.R.O.; Dales, N.; Andrews, P.C. Surfactant effects on protein structure examined by electrospray ionization mass spectrometry. Protein Science 1994, 3, 1975-1983. [CrossRef]
- Shieh, I.F.; Lee, C.-Y.; Shiea, J. Eliminating the Interferences from TRIS Buffer and SDS in Protein Analysis by Fused-Droplet Electrospray Ionization Mass Spectrometry. Journal of Proteome Research 2005, 4, 606-612. [CrossRef]
- Rogers, J.C.; Bomgarden, R.D. Sample preparation for mass spectrometry-based proteomics; from proteomes to peptides. In Modern Proteomics–Sample Preparation, Analysis and Practical Applications; Springer: 2016; pp. 43-62.
- Lebert, D.; Picard, G.; Beau-Larvor, C.; Troncy, L.; Lacheny, C.; Maynadier, B.; Low, W.; Mouz, N.; Brun, V.; Klinguer-Hamour, C. Absolute and multiplex quantification of antibodies in serum using PSAQ™ standards and LC-MS/MS. Bioanalysis 2015, 7, 1237-1251. [CrossRef]
- Martos, G.; Bedu, M.; Josephs, R.; Westwood, S.; Wielgosz, R. Quantification of SARS-CoV-2 monoclonal IgG mass fraction by isotope dilution mass spectrometry. Analytical and Bioanalytical Chemistry 2024, 416, 2423-2437. [CrossRef]
- Suttapitugsakul, S.; Xiao, H.; Smeekens, J.; Wu, R. Evaluation and optimization of reduction and alkylation methods to maximize peptide identification with MS-based proteomics. Molecular BioSystems 2017, 13, 2574-2582. [CrossRef]
- Müller, T.; Winter, D. Systematic evaluation of protein reduction and alkylation reveals massive unspecific side effects by iodine-containing reagents. Molecular & cellular proteomics 2017, 16, 1173-1187. [CrossRef]
- Kuznetsova, K.G.; Levitsky, L.I.; Pyatnitskiy, M.A.; Ilina, I.Y.; Bubis, J.A.; Solovyeva, E.M.; Zgoda, V.G.; Gorshkov, M.V.; Moshkovskii, S.A. Cysteine alkylation methods in shotgun proteomics and their possible effects on methionine residues. Journal of proteomics 2021, 231, 104022. [CrossRef]
- Hains, P.G.; Robinson, P.J. The impact of commonly used alkylating agents on artifactual peptide modification. Journal of proteome research 2017, 16, 3443-3447. [CrossRef]
- Geoghegan, K.F.; Hoth, L.R.; Tan, D.H.; Borzilleri, K.A.; Withka, J.M.; Boyd, J.G. Cyclization of N-terminal S-carbamoylmethylcysteine causing loss of 17 Da from peptides and extra peaks in peptide maps. Journal of proteome research 2002, 1, 181-187. [CrossRef]
- Switzar, L.; Giera, M.; Niessen, W.M. Protein digestion: an overview of the available techniques and recent developments. Journal of proteome research 2013, 12, 1067-1077. [CrossRef]
- Falck, D.; Jansen, B.C.; Plomp, R.; Reusch, D.; Haberger, M.; Wuhrer, M. Glycoforms of immunoglobulin G based biopharmaceuticals are differentially cleaved by trypsin due to the glycoform influence on higher-order structure. Journal of proteome research 2015, 14, 4019-4028. [CrossRef]
- Heissel, S.; Frederiksen, S.J.; Bunkenborg, J.; Højrup, P. Enhanced trypsin on a budget: Stabilization, purification and high-temperature application of inexpensive commercial trypsin for proteomics applications. PLOS ONE 2019, 14, e0218374. [CrossRef]
- Menneteau, T.; Saveliev, S.; Butré, C.I.; Rivera, A.K.G.; Urh, M.; Delobel, A. Addressing common challenges of biotherapeutic protein peptide mapping using recombinant trypsin. Journal of Pharmaceutical and Biomedical Analysis 2024, 243, 116124. [CrossRef]
- Reinders, L.M.; Klassen, M.D.; Teutenberg, T.; Jaeger, M.; Schmidt, T.C. Development of a multidimensional online method for the characterization and quantification of monoclonal antibodies using immobilized flow-through enzyme reactors. Analytical and Bioanalytical Chemistry 2021, 413, 7119-7128. [CrossRef]
- Glatter, T.; Ludwig, C.; Ahrné, E.; Aebersold, R.; Heck, A.J.R.; Schmidt, A. Large-Scale Quantitative Assessment of Different In-Solution Protein Digestion Protocols Reveals Superior Cleavage Efficiency of Tandem Lys-C/Trypsin Proteolysis over Trypsin Digestion. Journal of Proteome Research 2012, 11, 5145-5156. [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nature Protocols 2016, 11, 993-1006. [CrossRef]
- Trevisiol, S.; Ayoub, D.; Lesur, A.; Ancheva, L.; Gallien, S.; Domon, B. The use of proteases complementary to trypsin to probe isoforms and modifications. PROTEOMICS 2016, 16, 715-728. [CrossRef]
- Hao, P.; Ren, Y.; Datta, A.; Tam, J.P.; Sze, S.K. Evaluation of the effect of trypsin digestion buffers on artificial deamidation. Journal of proteome research 2015, 14, 1308-1314. [CrossRef]
- Sutherland, E.; Veth, T.S.; Riley, N.M. Revisiting the effect of trypsin digestion buffers on artificial deamidation. 2024. [CrossRef]
- Jiang, H.; Zeng, J.; Titsch, C.; Voronin, K.; Akinsanya, B.; Luo, L.; Shen, H.; Desai, D.D.; Allentoff, A.; Aubry, A.-F.; et al. Fully Validated LC-MS/MS Assay for the Simultaneous Quantitation of Coadministered Therapeutic Antibodies in Cynomolgus Monkey Serum. Analytical Chemistry 2013, 85, 9859-9867. [CrossRef]
- Ren, D.; Pipes, G.D.; Liu, D.; Shih, L.-Y.; Nichols, A.C.; Treuheit, M.J.; Brems, D.N.; Bondarenko, P.V. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Analytical biochemistry 2009, 392, 12-21. [CrossRef]
- Liu, H.; Ponniah, G.; Neill, A.; Patel, R.; Andrien, B. Accurate determination of protein methionine oxidation by stable isotope labeling and LC-MS analysis. Analytical chemistry 2013, 85, 11705-11709. [CrossRef]
- Yang, J.; Gao, Z.; Ren, X.; Sheng, J.; Xu, P.; Chang, C.; Fu, Y. DeepDigest: Prediction of Protein Proteolytic Digestion with Deep Learning. Analytical Chemistry 2021, 93, 6094-6103. [CrossRef]
- Gupta, S.; Jiskoot, W.; Schöneich, C.; Rathore, A.S. Oxidation and deamidation of monoclonal antibody products: potential impact on stability, biological activity, and efficacy. Journal of Pharmaceutical Sciences 2022, 111, 903-918. [CrossRef]
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Buchanan, T.; Guender, M.; Kristensen, D.B.; Sloth, T.M.; Ørgaard, M.; Cook, K.; Bones, J. Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Analytical and Bioanalytical Chemistry 2020, 412, 6833-6848. [CrossRef]
- Kuzyk, M.A.; Parker, C.E.; Domanski, D.; Borchers, C.H. Development of MRM-based assays for the absolute quantitation of plasma proteins. Methods in Molecular Biology (Clifton, N.J.) 2013, 53-82. [CrossRef]
- Liebler, D.C.; Zimmerman, L.J. Targeted quantitation of proteins by mass spectrometry. Biochemistry 2013, 52, 3797-3806. [CrossRef]
- Schmidt, C.; Urlaub, H. Absolute quantification of proteins using standard peptides and multiple reaction monitoring. Quantitative Methods in Proteomics 2012, 249-265. [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. In Biocomputing 2002; World Scientific: 2001; pp. 310-322.
- Pugalenthi, G.; Nithya, V.; Chou, K.-C.; Archunan, G. Nglyc: a random forest method for prediction of N-glycosylation sites in eukaryotic protein sequence. Protein and Peptide Letters 2020, 27, 178-186. [CrossRef]
- Pakhrin, S.C.; Aoki-Kinoshita, K.F.; Caragea, D.; Kc, D.B. DeepNGlyPred: a deep neural network-based approach for human N-linked glycosylation site prediction. Molecules 2021, 26, 7314. [CrossRef]
- Jian, W.; Kang, L.; Burton, L.; Weng, N. A workflow for absolute quantitation of large therapeutic proteins in biological samples at intact level using LC-HRMS. Bioanalysis 2016, 8, 1679-1691. [CrossRef]
- Lanshoeft, C.; Cianférani, S.; Heudi, O. Generic Hybrid Ligand Binding Assay Liquid Chromatography High-Resolution Mass Spectrometry-Based Workflow for Multiplexed Human Immunoglobulin G1 Quantification at the Intact Protein Level: Application to Preclinical Pharmacokinetic Studies. Analytical Chemistry 2017, 89, 2628-2635. [CrossRef]
- Cong, Y.; Zhang, Z.; Zhang, S.; Hu, L.; Gu, J. Quantitative MS analysis of therapeutic mAbs and their glycosylation for pharmacokinetics study. PROTEOMICS - Clinical Applications 2016, 10, 303-314. [CrossRef]
- Liu, S.; Liu, X. IgG N-glycans. Advances in Clinical Chemistry 2021, 105, 1-47. [CrossRef]
- van den Broek, I.; van Dongen, W.D. LC–MS-based quantification of intact proteins: perspective for clinical and bioanalytical applications. Bioanalysis 2015, 7, 1943-1958. [CrossRef]
- Rosati, S.; Yang, Y.; Barendregt, A.; Heck, A.J.R. Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nature Protocols 2014, 9, 967-976. [CrossRef]
- Xu, K.; Liu, L.; Saad, O.M.; Baudys, J.; Williams, L.; Leipold, D.; Shen, B.; Raab, H.; Junutula, J.R.; Kim, A. Characterization of intact antibody–drug conjugates from plasma/serum in vivo by affinity capture capillary liquid chromatography–mass spectrometry. Analytical biochemistry 2011, 412, 56-66. [CrossRef]
- Jin, W.; Burton, L.; Moore, I. LC–HRMS quantitation of intact antibody drug conjugate trastuzumab emtansine from rat plasma. Bioanalysis 2018, 10, 851-862. [CrossRef]
- Zhao, Y.; Gu, H.; Zheng, N.; Zeng, J. Critical considerations for immunocapture enrichment LC–MS bioanalysis of protein therapeutics and biomarkers. Bioanalysis 2018, 10, 987-995. [CrossRef]
- Kang, L.; Weng, N.; Jian, W. LC–MS bioanalysis of intact proteins and peptides. Biomedical Chromatography 2020, 34, e4633. [CrossRef]
- Zhang, L.; Vasicek, L.A.; Hsieh, S.; Zhang, S.; Bateman, K.P.; Henion, J. Top-down LC–MS quantitation of intact denatured and native monoclonal antibodies in biological samples. Bioanalysis 2018, 10, 1039-1054. [CrossRef]
- Song, Y.E.; Lebert, D.; Guillaubez, J.-V.; Samra, S.; Goucher, E.; Hart, B. Streamlined workflow for absolute quantitation of therapeutic monoclonal antibodies using Promise Proteomics mAbXmise kits and a TSQ Altis Plus mass spectrometer. Available online: https://promise-proteomics.com/wp-content/uploads/2023/06/tn-001753-cl-clinical-altis-mabs-tn001753-na-en.pdf (accessed on 07.08.2024).
- Rutherfurd, S.M.; Gilani, G.S. Amino Acid Analysis. Current Protocols in Protein Science 2009, 58. [CrossRef]
- Rutherfurd, S.M.; Dunn, B.M. Quantitative Amino Acid Analysis. Current Protocols in Protein Science 2011, 63. [CrossRef]
- Josephs, R.D.; Martos, G.; Li, M.; Wu, L.; Melanson, J.E.; Quaglia, M.; Beltrão, P.J.; Prevoo-Franzsen, D.; Boeuf, A.; Delatour, V. Establishment of measurement traceability for peptide and protein quantification through rigorous purity assessment—a review. Metrologia 2019, 56, 044006. [CrossRef]
- Violi, J.P.; Bishop, D.P.; Padula, M.P.; Steele, J.R.; Rodgers, K.J. Considerations for amino acid analysis by liquid chromatography-tandem mass spectrometry: A tutorial review. TrAC Trends in Analytical Chemistry 2020, 131, 116018. [CrossRef]
- Kato, M.; Kato, H.; Eyama, S.; Takatsu, A. Application of amino acid analysis using hydrophilic interaction liquid chromatography coupled with isotope dilution mass spectrometry for peptide and protein quantification. Journal of Chromatography B 2009, 877, 3059-3064. [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; Schrag, J.D.; Melanson, J.E. Characterization of a SARS-CoV-2 spike protein reference material. Analytical and Bioanalytical Chemistry 2022, 414, 3561-3569. [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; L’Abbé, D.; Stuible, M.; Durocher, Y.; Melanson, J.E. Production and Characterization of a SARS-CoV-2 Nucleocapsid Protein Reference Material. ACS Measurement Science Au 2022, 2, 620-628. [CrossRef]
- Louwagie, M.; Kieffer-Jaquinod, S.; Dupierris, V.; Couté, Y.; Bruley, C.; Garin, J.; Dupuis, A.; Jaquinod, M.; Brun, V. Introducing AAA-MS, a Rapid and Sensitive Method for Amino Acid Analysis Using Isotope Dilution and High-Resolution Mass Spectrometry. Journal of Proteome Research 2012, 11, 3929-3936. [CrossRef]
- Jeong, J.-S.; Lim, H.-M.; Kim, S.-K.; Ku, H.-K.; Oh, K.-H.; Park, S.-R. Quantification of human growth hormone by amino acid composition analysis using isotope dilution liquid-chromatography tandem mass spectrometry. Journal of Chromatography a 2011, 1218, 6596-6602. [CrossRef]
- Mi, W.; Josephs, R.; Melanson, J.; Dai, X.; Wang, Y.; Zhai, R.; Chu, Z.; Fang, X.; Thibeault, M.; Stocks, B. PAWG Pilot Study on Quantification of SARS-CoV-2 Monoclonal Antibody-Part 1. Metrologia 2022, 59, 08001. [CrossRef]
- Stocks, B.B.; Thibeault, M.-P.; L’Abbé, D.; Umer, M.; Liu, Y.; Stuible, M.; Durocher, Y.; Melanson, J.E. Characterization of biotinylated human ACE2 and SARS-CoV-2 Omicron BA. 4/5 spike protein reference materials. Analytical and Bioanalytical Chemistry 2024, 1-12. [CrossRef]
- Catalogue: SIL-mAbs for targeted LC-MS quantification. Available online: https://promise-proteomics.com/wp-content/uploads/2024/01/Catalogue-SIL-mAbs-2024.pdf (accessed on 07.08.2024).
- Picard, G.; Lebert, D.; Louwagie, M.; Adrait, A.; Huillet, C.; Vandenesch, F.; Bruley, C.; Garin, J.; Jaquinod, M.; Brun, V. PSAQ™ standards for accurate MS-based quantification of proteins: from the concept to biomedical applications. Journal of Mass Spectrometry 2012, 47, 1353-1363. [CrossRef]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Molecular & Cellular Proteomics 2002, 1, 376-386. [CrossRef]
- Osaki, F.; Tabata, K.; Oe, T. Quantitative LC/ESI-SRM/MS of antibody biopharmaceuticals: use of a homologous antibody as an internal standard and three-step method development. Analytical and Bioanalytical Chemistry 2017, 409, 5523-5532. [CrossRef]
- Smit, N.P.M.; Ruhaak, L.R.; Romijn, F.P.H.T.M.; Pieterse, M.M.; Van Der Burgt, Y.E.M.; Cobbaert, C.M. The Time Has Come for Quantitative Protein Mass Spectrometry Tests That Target Unmet Clinical Needs. Journal of the American Society for Mass Spectrometry 2021, 32, 636-647. [CrossRef]
- Lee, H.; Lee, J. Peptide purity assignment for antibody quantification by combining isotope dilution mass spectrometry and liquid chromatography. Bulletin of the Korean Chemical Society 2022, 43, 704-713. [CrossRef]
- Burkitt, W.I.; Pritchard, C.; Arsene, C.; Henrion, A.; Bunk, D.; O’Connor, G. Toward Systeme International d’Unite-traceable protein quantification: from amino acids to proteins. Analytical biochemistry 2008, 376, 242-251. [CrossRef]
- Benesova, E.; Vidova, V.; Spacil, Z. A comparative study of synthetic winged peptides for absolute protein quantification. Scientific Reports 2021, 11. [CrossRef]
- Li, H.; Ortiz, R.; Tran, L.; Hall, M.; Spahr, C.; Walker, K.; Laudemann, J.; Miller, S.; Salimi-Moosavi, H.; Lee, J.W. General LC-MS/MS Method Approach to Quantify Therapeutic Monoclonal Antibodies Using a Common Whole Antibody Internal Standard with Application to Preclinical Studies. Analytical Chemistry 2012, 84, 1267-1273. [CrossRef]
- Bronsema, K.J.; Bischoff, R.; van de Merbel, N.C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. Journal of Chromatography B 2012, 893, 1-14. [CrossRef]
- Furlong, M.T.; Ouyang, Z.; Wu, S.; Tamura, J.; Olah, T.; Tymiak, A.; Jemal, M. A universal surrogate peptide to enable LC-MS/MS bioanalysis of a diversity of human monoclonal antibody and human Fc-fusion protein drug candidates in pre-clinical animal studies. Biomedical Chromatography 2012, 26, 1024-1032. [CrossRef]
- Furlong, M.T. Generic Peptide Strategies for LC–MS/MS Bioanalysis of Human Monoclonal Antibody Drugs and Drug Candidates. Protein Analysis using Mass Spectrometry: Accelerating Protein Biotherapeutics from Lab to Patient 2017, 161-181. [CrossRef]
- Administration, F.a.D. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry (accessed on 07.08.2024).
- Agency, E.M. ICH guideline M10 on bioanalytical method validation and study sample analysis. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on 07.08.2024).
- Vidova, V.; Spacil, Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Analytica chimica acta 2017, 964, 7-23. [CrossRef]
- Ranbaduge, N.; Yu, Y.Q. A Streamlined Compliant Ready Workflow for Peptide-Based Multi-Attribute Method (MAM). Available online: https://www.waters.com/content/dam/waters/en/app-notes/2020/720007094/720007094-zh_tw.pdf (accessed on.
- Yun W. Alelyunas, H.S., Mark D. Wrona, Weibin Chen Rapid, Sensitive, and Routine Intact mAb Quantification using a Compact Tof HRMS Platform. Available online: https://lcms.cz/labrulez-bucket-strapi-h3hsga3/2019asms_alelyunas_intactproteinquan_659b6f83aa/2019asms_alelyunas_intactproteinquan.pdf (accessed on 07.08.2024).
- Millán-Martín, S.; Jakes, C.; Carillo, S.; Bones, J. Multi-Attribute Method (MAM) Analytical Workflow for Biotherapeutic Protein Characterization from Process Development to QC. Current Protocols 2023, 3, e927. [CrossRef]
- Schneck, N.A.; Mehl, J.T.; Kellie, J.F. Protein LC-MS Tools for the Next Generation of Biotherapeutic Analyses from Preclinical and Clinical Serum. Journal of the American Society for Mass Spectrometry 2023, 34, 1837-1846. [CrossRef]
- Kiyonami, R.; Schoen, A.; Prakash, A.; Nguyen, H.; Peterman, S.; Selevsek, N.; Zabrouskov, V.; Huhmer, A.; Domon, B. Rapid assay development and refinement for targeted protein quantitation using an intelligent SRM (iSRM) workflow. Available online: https://tools.thermofisher.com/content/sfs/brochures/AN468_63139_Vantage_Prot(1).pdf (accessed on 07.08.2024).
- Kiyonami, R.; Zeller, M.; Zabrouskov, V. Quantifying peptides in complex mixtures with high sensitivity and precision using a targeted approach with a hybrid linear ion trap-orbitrap mass spectrometer. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-557-LC-MS-Peptides-Complex-Mixtures-AN63499-EN.pdf (accessed on 07.08.2024).
- Colangelo, C.M.; Chung, L.; Bruce, C.; Cheung, K.-H. Review of software tools for design and analysis of large scale MRM proteomic datasets. Methods 2013, 61, 287-298. [CrossRef]
- Desiere, F.; Deutsch, E.W.; King, N.L.; Nesvizhskii, A.I.; Mallick, P.; Eng, J.; Chen, S.; Eddes, J.; Loevenich, S.N.; Aebersold, R. The peptideatlas project. Nucleic acids research 2006, 34, D655-D658. [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966-968. [CrossRef]
- Gessulat, S.; Schmidt, T.; Zolg, D.P.; Samaras, P.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Rechenberger, J.; Delanghe, B.; Huhmer, A. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nature methods 2019, 16, 509-518.
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nature biotechnology 2008, 26, 1367-1372. [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nature protocols 2009, 4, 698-705. [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature protocols 2016, 11, 2301-2319.
- Röst, H.L.; Sachsenberg, T.; Aiche, S.; Bielow, C.; Weisser, H.; Aicheler, F.; Andreotti, S.; Ehrlich, H.-C.; Gutenbrunner, P.; Kenar, E. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nature methods 2016, 13, 741-748. [CrossRef]
- Pfeuffer, J.; Bielow, C.; Wein, S.; Jeong, K.; Netz, E.; Walter, A.; Alka, O.; Nilse, L.; Colaianni, P.D.; McCloskey, D. OpenMS 3 enables reproducible analysis of large-scale mass spectrometry data. Nature methods 2024, 21, 365-367. [CrossRef]
- Tsugawa, H.; Kanazawa, M.; Ogiwara, A.; Arita, M. MRMPROBS suite for metabolomics using large-scale MRM assays. Bioinformatics 2014, 30, 2379-2380. [CrossRef]
- Valot, B.; Langella, O.; Nano, E.; Zivy, M. MassChroQ: a versatile tool for mass spectrometry quantification. Proteomics 2011, 11, 3572-3577. [CrossRef]
- Cai, Y.; Weng, K.; Guo, Y.; Peng, J.; Zhu, Z.-J. An integrated targeted metabolomic platform for high-throughput metabolite profiling and automated data processing. Metabolomics 2015, 11, 1575-1586. [CrossRef]
- Team, R.C. R: A language and environment for statistical computing. Available online: https://www.R-project.org/ (accessed on 07.08.2024).
- Choi, M.; Carver, J.; Chiva, C.; Tzouros, M.; Huang, T.; Tsai, T.-H.; Pullman, B.; Bernhardt, O.M.; Hüttenhain, R.; Teo, G.C. MassIVE. quant: a community resource of quantitative mass spectrometry–based proteomics datasets. Nature methods 2020, 17, 981-984. [CrossRef]
- Dai, C.; Pfeuffer, J.; Wang, H.; Zheng, P.; Käll, L.; Sachsenberg, T.; Demichev, V.; Bai, M.; Kohlbacher, O.; Perez-Riverol, Y. quantms: a cloud-based pipeline for quantitative proteomics enables the reanalysis of public proteomics data. Nature Methods 2024, 1-5. [CrossRef]
- Wang, R.; Jiang, H.; Lu, M.; Tong, J.; An, S.; Wang, J.; Yu, C. MRMPro: a web-based tool to improve the speed of manual calibration for multiple reaction monitoring data analysis by mass spectrometry. BMC bioinformatics 2024, 25, 60. [CrossRef]
- Mi, W.; Josephs, R.; Melanson, J.; Dai, X.; Wang, Y.; Zhai, R.; Chu, Z.; Fang, X.; Thibeault, M.; Stocks, B. PAWG pilot study on quantification of SARS-CoV-2 monoclonal antibody-part 2. Metrologia 2023, 60, 08016. [CrossRef]
- Miller, W.G.; Greenberg, N.; Panteghini, M.; Budd, J.R.; Johansen, J.V. Guidance on which calibrators in a metrologically traceable calibration hierarchy must be commutable with clinical samples. Clinical Chemistry 2023, 69, 228-238. [CrossRef]
- Diederiks, N.M.; van der Burgt, Y.E.; Ruhaak, L.R.; Cobbaert, C.M. Developing an SI-traceable Lp (a) reference measurement system: a pilgrimage to selective and accurate apo (a) quantification. Critical Reviews in Clinical Laboratory Sciences 2023, 60, 483-501. [CrossRef]
- Ruhaak, L.R.; Romijn, F.P.; Begcevic Brkovic, I.; Kuklenyik, Z.; Dittrich, J.; Ceglarek, U.; Hoofnagle, A.N.; Althaus, H.; Angles-Cano, E.; Coassin, S. Development of an LC-MRM-MS-based candidate reference measurement procedure for standardization of serum apolipoprotein (a) tests. Clinical Chemistry 2023, 69, 251-261. [CrossRef]
- Schiel, J.E.; Mire-Sluis, A.; Davis, D. Monoclonal antibody therapeutics: the need for biopharmaceutical reference materials. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1. Monoclonal Antibody Therapeutics: Structure, Function, and Regulatory Space; ACS Publications: 2014; pp. 1-34.
- Schiel, J.E.; Turner, A. The NISTmAb Reference Material 8671 lifecycle management and quality plan. Analytical and bioanalytical chemistry 2018, 410, 2067-2078. [CrossRef]
- Kinumi, T.; Saikusa, K.; Kato, M.; Kojima, R.; Igarashi, C.; Noda, N.; Honda, S. Characterization and Value Assignment of a Monoclonal Antibody Reference Material, NMIJ RM 6208a, AIST-MAB. Frontiers in Molecular Biosciences 2022, 9, 842041. [CrossRef]
- Dong, L.; Zhang, Y.; Fu, B.; Swart, C.; Jiang, H.; Liu, Y.; Huggett, J.; Wielgosz, R.; Niu, C.; Li, Q. Reliable biological and multi-omics research through biometrology. Analytical and Bioanalytical Chemistry 2024, 1-19. [CrossRef]
- Shuford, C.M.; Walters, J.J.; Holland, P.M.; Sreenivasan, U.; Askari, N.; Ray, K.; Grant, R.P. Absolute protein quantification by mass spectrometry: not as simple as advertised. Analytical chemistry 2017, 89, 7406-7415. [CrossRef]
- Zhong, X.; Chen, H.; Zare, R.N. Ultrafast enzymatic digestion of proteins by microdroplet mass spectrometry. Nature communications 2020, 11, 1049. [CrossRef]
- Xiao, T.; Li, Z.; Xing, X.; He, F.; Huang, J.; Xue, D. Improving the activity and thermal stability of trypsin by the rational design. Process Biochemistry 2023, 130, 227-235. [CrossRef]
- Guo, C.; Liu, Y.; Yu, H.; Du, K.; Gan, Y.; Huang, H. A novel strategy for thermostability improvement of trypsin based on N-glycosylation within the Ω-loop region. Journal of Microbiology and Biotechnology 2016, 26, 1163-1172. [CrossRef]
- Narumi, R.; Masuda, K.; Tomonaga, T.; Adachi, J.; Ueda, H.R.; Shimizu, Y. Cell-free synthesis of stable isotope-labeled internal standards for targeted quantitative proteomics. Synthetic and Systems Biotechnology 2018, 3, 97-104. [CrossRef]
Figure 2.
Overview of MS-based antibody quantification strategies of pure or matrix-containing antibody samples. Proteolytic enzymes cleave the antibody sequence at specific positions, such as the C-terminal bond of lysine and arginine (trypsin), releasing defined peptide fragments. The treatment of antibodies with acids under high temperatures enables quantification of free amino acids.
Figure 2.
Overview of MS-based antibody quantification strategies of pure or matrix-containing antibody samples. Proteolytic enzymes cleave the antibody sequence at specific positions, such as the C-terminal bond of lysine and arginine (trypsin), releasing defined peptide fragments. The treatment of antibodies with acids under high temperatures enables quantification of free amino acids.
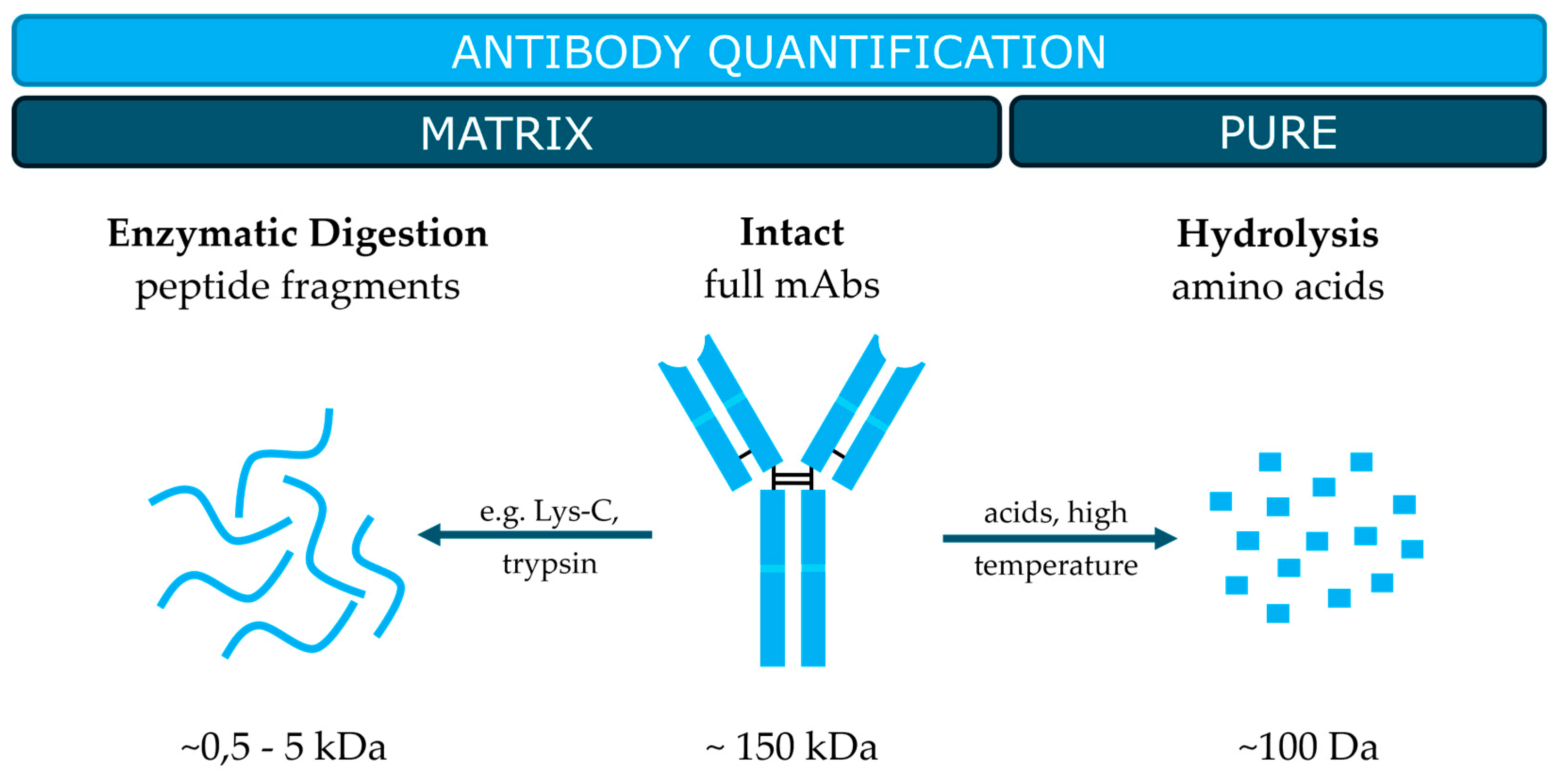
Figure 3.
Cleavage sites of commonly used proteases for bottom-up proteomics.

Table 1.
Top 5 best-selling recombinant antibodies approved for therapeutic application.1 Yearly turn-over in 2023 [21]. 2Recently published peptide-based quantification methods for the respective mAb and the selected signature peptides.
Table 1.
Top 5 best-selling recombinant antibodies approved for therapeutic application.1 Yearly turn-over in 2023 [21]. 2Recently published peptide-based quantification methods for the respective mAb and the selected signature peptides.
| Generic name | Antibody subclass | Year of approval |
Target | Sales1 (Billion USD) |
Signature peptides2 |
|---|---|---|---|---|---|
| Pembrolizumab | IgG4κ | 2021 | Programmed cell death protein 1 (PD-1) | 25.0 | ASGYTFTNYYMYWVR [28] DLPLTFGGGTK [28,29] VTLTTDSSTTTAYMELK [30] |
| Adalimumab | IgG1κ | 2002 | Tumor necrosis factor alpha (TNF-α) |
14.4 | APYTFGQGTK [31,32] |
| Ustekinumab | IgG1κ | 2009 | Interleukin (IL)-12 and IL-23 | 10.9 | PGQGYFDFWGQGTLVTVSSSSTK [33] GLDWIGIMSPVDSDIR [29,33] |
| Daratumumab | IgG1κ | 2016 | Hydrolase CD38 | 9.7 | SNWPPTFGQGTK [34] LLIYDASNR [35] |
| Nivolumab | IgG4κ | 2014 | PD-1 | 9.0 | ASGITFSNSGMHWVR [29,30,36,37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



