
Submitted:
19 December 2024
Posted:
20 December 2024
You are already at the latest version
Abstract
The study of mitochondrial dysfunction has become increasingly pivotal in elucidating the pathophysiology of various cerebral pathologies, particularly neurodegenerative disorders. Mitochondria are essential for cellular energy metabolism, regulation of reactive oxygen species (ROS), calcium homeostasis, and the execution of apoptotic processes. Disruptions in mitochondrial function, driven by factors such as oxidative stress, excitotoxicity, and altered ion balance, lead to neuronal death and contribute to cognitive impairments in several brain diseases. Mitochondrial dysfunction can arise from genetic mutations, ischemic events, hypoxia, and other environmental factors. This article highlights the critical role of mitochondrial dysfunction in the progression of neurodegenerative diseases and discusses the need for targeted therapeutic strategies to attenuate cellular damage, restore mitochondrial function, and enhance neuroprotection.
Keywords:
mitochondrial dysfunction
; ROS
; cerebral ischemia
; HIF-1
; HSP 70
1. Introduction
The relevance of the study of various types of cerebral pathology and the development of methods for their treatment does not require detailed justification [1]. The World Health Organization defines human death as the death of the brain, which in life controls all the most important functions. In terms of prevalence and mortality, brain diseases rank third among diseases of the population of industrialized countries, lead not only to a decrease in life expectancy of the population, but also limit the social activity of a person due to the development of cognitive deficit, a decrease in the individual's ability to think, learn, adequately perceive information and make decisions [2,3]. In brain diseases of destructive and degenerative genesis, mitochondrial respiratory chain, energy metabolism, ionic homeostasis of the cell with increased content of calcium ions, development of glutamate excitotoxicity and damaging effect of nitrosative and oxidative stress, initiation of neuroapoptosis and cell death occur [4]. The primary source of reactive oxygen species (ROS) is mitochondria, which play a key role in the cell's energy supply [5]. Nowadays there is a generalized concept of “mitochondrial dysfunction”. This is a typical pathological process that has no etiologic and nosologic specificity. The development of mitochondrial dysfunction eventually leads to neuron death [6]. We can speak about mitochondrial dysfunction as a new pathobiochemical mechanism of neurodegenerative disorders of a wide spectrum. All of the above is the rationale for the search for highly effective cerebroprotective drugs capable of preventing negative processes of mitochondrial dysfunction in the cell, thereby having a cerebroprotective effect [7]. Currently, energy-tropic drugs such as coenzyme Q10, carnitine, B vitamins, succinic acid derivatives, etc. are being tried to correct mitochondrial dysfunction [6,8]. However, the rational basis for their use is poorly developed, and effective approaches are often underutilized or ineffective ones are overestimated. Medications are applied chaotically, without sufficient knowledge of their potential and characteristics, and without planning a treatment strategy from the perspective of appropriateness. Moreover, in cases of established mitochondrial dysfunction and the activation of apoptotic processes, these drugs are largely ineffective as they cannot regulate the subtle mechanisms of energy metabolism in which they act as intermediates.
Another approach to correcting mitochondrial dysfunction is also being considered: the use of thiol antioxidants. These compete with the SH-groups of the cysteine-dependent site of the mitochondrial inner membrane protein (ATP/ADP antiporter) for ROS and peroxynitrite, forming stable complexes with the latter. This prevents the opening of the mitochondrial pore under conditions of oxidative and nitrosative stress.
Particularly interesting and deserving of special attention is the use of drugs that act as ligands for neuropeptide receptors. These can regulate apoptosis, the expression of transcription factors, the synthesis of enzymes that regenerate mitochondrial DNA, and enzymes that catalyze energy reactions. Recently, significant efforts have been directed toward identifying highly effective neuroprotectors among neuropeptides.
The above highlights the scientific appeal of studying mitochondrial dysfunction in neurons, making the development of new approaches to pharmacological correction highly relevant and promising from the standpoint of comprehensive neuroprotection [6,9,10].
1.1. The Role of Mitochondria in the Energy Metabolism of the Brain
The relationship between the structure and function of mitochondria remains a central focus of attention for a wide range of researchers. In recent years, revolutionary developments have taken place and are continuing in mitochondrial biology. Traditional views on the role of mitochondria in the cell have undergone significant revisions. Until relatively recently, our understanding of mitochondria was limited to their function as the cell's energy stations, and the role of mitochondria in the development of pathology was confined to energy supply disruption, with all ultrastructural changes in mitochondria being viewed exclusively from this perspective. However, many ultrastructural states of mitochondria cannot be explained within such frameworks. Today, there is no doubt about the crucial significance of mitochondria for the life of eukaryotic cells. The dominant role of these organelles in ATP production, the execution of programmed cell death processes, their involvement in the generation of reactive oxygen species (ROS), and the storage of calcium ions all determine their participation in the development of many pathological processes [6,11].
The brain has extremely high metabolic needs. It uses approximately 20% of the body's total oxygen and glucose consumption with only 2% of its body weight. About 70% of estimated energy expenditure is used to support neuronal signaling, including resting potentials, action potentials, postsynaptic receptor activation, glutamate cycling, and postsynaptic Ca2+ signaling , while the remainder goes to nonsignaling activities such as biomacromolecule trafficking, axonal transport, mitochondrial proton leakage, and actin cytoskeleton remodeling [12,13,14].
Neurons exhibit the majority of energy consumption. They generate ATP primarily in mitochondria via oxidative phosphorylation, with a small portion of ATP from aerobic glycolysis in the cytoplasm. Astrocytes are highly glycolytic and convert glucose to lactate with low oxygen consumption; lactate is then delivered to neurons for complete oxidation. This process largely supports the energy requirements of neurons by supplying metabolic substrates [15,16].
Oligodendrocytes also obtain ATP mainly through aerobic glycolysis. They use lactate for their own energy needs and also supply neighboring axons with lactate. Microglia are mainly nourished by oxidative phosphorylation, but are metabolically reprogrammed to a phenotype dominated by aerobic glycolysis under certain neurological circumstances [17,18,19]. Astrocytes have low mitochondrial activity of oxidative phosphorylation processes; this metabolic mode of astrocytes is essential for brain lipid homeostasis. Aberrant astrocytic oxidative phosphorylation process can cause accumulation of lipid droplets with subsequent development of neurodegeneration [20].
1.2. Concept of Mitochondrial Dysfunction
To date, on the basis of experimental studies, the concept of mitochondrial dysfunction, its formation, molecular, biochemical and ultrastructural features has been formulated [6,21,22,23].
The development of mitochondrial dysfunction leads to the disruption of neurotransmitter reuptake (catecholamines, dopamine, serotonin), ion transport, impulse generation and conduction, de novo protein synthesis, translation and transcription processes; "parasitic" energy-producing reactions are activated, resulting in a significant depletion of the neuronal cell's energy reserves. Additionally, under the influence of reactive oxygen species (ROS), particularly the hydroxyl radical, mitochondrial pores are opened, leading to the expression and release of pro-apoptotic proteins into the cytosol. The opening of these pores occurs due to the oxidation of thiol groups in the cysteine-dependent region of the inner mitochondrial membrane protein (ATP/ADP antiporter), transforming it into a permeable nonspecific channel-pore. Today, the primary mechanism for this process is recognized as the formation of mitochondrial apoptotic pores and pores of increased permeability (РТP – permeability transition pores) [6,24,25,26,27].
The opening of the giant pore makes the mitochondrial membranes permeable to dissolved substances with a molecular mass of 1500 Da and more. This disrupts mitochondrial metabolism, halting the synthesis of mitochondrial proteins and the import of proteins synthesized in the cytosol. Oxidative phosphorylation becomes uncoupled, and ATP synthesis stops. Hyperproduction of O2- begins, and reducing equivalents are depleted. The opening of the pore transforms the mitochondria from "power plants" into "furnaces" for oxidation substrates without ATP production [28,29].
It was found that interaction of mitochondrial structures with active derivatives of NO and ROS, Ca2+ “overbreathing” , reduction of intramitochondrial GSH enhances pore opening and release of apoptogenic proteins from damaged mitochondria [30,31]. In this context, the role of one of the neurotrophic factors, tumor necrosis factor-α (TNF-α), with which the opening of pores in mitochondria, the subsequent disruption of their membranes and the development of mitoptosis are associated, is significant [32].
Thus, we can speak about mitochondrial dysfunction as a new pathobiochemical mechanism of neurodegenerative disorders of a wide spectrum. At present, two types of mitochodrial dysfunction are distinguished - primary as a consequence of congenital genetic defect and secondary, arising under the influence of various factors: hypoxia, ischemia, oxidative and nitrosative stress, expression of proinflammatory cytokines. In modern medicine, the doctrine of polysystemic disorders of cellular energy exchange, the so-called mitochondrial pathology, or mitochondrial dysfunction, occupies an increasingly important position [6].
A key area of this section of medicine is hereditary syndromes based on mutations of genes responsible for mitochondrial proteins (Kearns-Sayre, MELAS, MERRF, Pearson, Barth, etc.) [33].
However, the class of conditions characterized by mitochondrial dysfunction is by no means limited to these primary mitochondrial dysfunctions. A huge number of diseases include disorders of cellular energy metabolism - secondary mitochondrial dysfunctions as important links in pathogenesis. Among them: intracerebral hemorrhage, epileptogenic seizures, localized thermal brain damage, neurodegenerative disorders, transient cerebral ischemia, chronic fatigue syndrome, migraines, cardiomyopathies, alcoholic encephalopathies, senile dementia, neuroinfections, cardiomyopathies, glycogenoses, connective tissue diseases, diabetes, rickets, tubulopathies, pancytopenia, hypoparathyroidism, liver failure, and many others (Figure 1) [6].
The study of these disorders is of particular importance for practical medicine due to the availability of effective therapeutic correction methods. However, it should be noted that the range of pathological disruptions in cellular energy metabolism is extremely wide (damages to various links in the Krebs cycle, respiratory chain, beta-oxidation, etc.). Mitochondrial dysfunction is closely associated with the hyperexpression of early genes, such as c-fos. In conditions of reactive oxygen species (ROS) hyperproduction by the neurochemical and bioenergetic systems of the brain during brain ischemia, as well as in several other neurodestructive pathological processes, there is an activation of the expression of redox-sensitive genes, many of which are essential for protecting cells from the toxic effects of oxidative stress [34,35,36].
Thus, under normal oxygen concentration in the surrounding cell environment (normoxia), the activation of JunB and ATF-2 transcription factors mainly occurs under the action of ROS, while under oxidative stress, the activation of c-Jun and c-fos factors predominates. The activation of these specific transcription factors under conditions of ROS hyperproduction is explained by the fact that JunB and c-fos contain cysteine residues (Cys252, Cys54, Cys61) in their DNA-binding domains that are highly sensitive to ROS. Oxidation of their SH-groups leads to the reverse inactivation of AR-1 and NF-kB [6,37,38].
In addition, c-fos protein is directly involved in the process of mitochondrial DNA fragmentation and initiation of apoptotic neuronal cell death. c-fos is responsible for NO hyperproduction in neurodegenerative diseases through iNOS activation. c-fos is one of the main nuclear targets for signaling regulation of cell growth and transformation, and is involved in many cellular functions, including cell proliferation and differentiation processes [39,40,41].
It is currently known that the main manifestations of mitochondrial dysfunction are a decrease in ATP levels in the cell, activation of cell death mechanisms, and the production of ROS by the mitochondria. It is known that during the functioning of the mitochondrial respiratory chain, small amounts of superoxide radical (O•–2) are formed as a byproduct of the respiratory complexes [42,43].
2. Molecular Mechanisms Behind Mitochondrial Dysfunction
2.1. Markers of Mitochondrial Dysfunction
One important mitochondrial control system that has recently been recognized is the sirtuins. These conserved class III histone deacetylases possess a catalytic nicotine-adenine dinucleotide + (NAD+) binding domain and play an important role in the regulation of both inflammation and metabolism [44,45].
In mitochondria, sirtuins utilize NAD+ as a sensor of mitochondrial energy status, with SIRT3 enhancing oxidative stress control by increasing SOD levels, SIRT 4 enhancing the ATP/ADP transporter, and SIRT 5 enhancing urea cycle function [46,47].
As a marker of mitochondrial dysfunction we use AMA-M2 - antimitochondrial antibodies directed to different proteins of the inner membrane of mitochondria. M2 is directed to the pyruvate dehydrogenase complex, and M3 to the malate dehydrogenase complex [48,49].
Also used as a marker of mitochondrial dysfunction is Cytochrome C, which is synthesized as apocytochrome C and enters the mitochondrion where it binds to the inner surface of the membrane and then it exits into the cytoplasm through channels that are opened for it by Bcl-2 family proteins [50,51,52].
Biochemical markers of mitochondrial dysfunction such as lactate and pyruvate are widely recognized. Pyruvate is a product of glycolysis, the pathway that metabolizes glucose for fuel, and is the first step in mitochondrial metabolism of carbohydrates. Pyruvate enters the tricarboxylic acid cycle, one of the main mitochondrial metabolic pathways. If there is mitochondrial dysfunction, the tricarboxylic acid cycle slows down, leading to pyruvate accumulation. As pyruvate accumulates, it is diverted to other pathways for metabolism, specifically lactate and alanine. Thus, when mitochondria become dysfunctional, three metabolic biomarkers can rise in the blood: pyruvate, lactate, and alanine. In fact, lactate and pyruvate are used in the diagnostic criteria for mitochondrial disease. The two ratios are also used to monitor mitochondrial function. The lactate to pyruvate ratio is elevated when lactate is overproduced from pyruvate. Another lesser-known ratio is the alanine-to-lysine ratio. Lysine is produced as a product of acetyl-CoA, the first step of the tricarboxylic acid cycle. Thus, when the tricarboxylic acid cycle slows down, less lysine and more alanine is produced, raising the alanine-to-lysine ratio to over 2.5 [53,54,55,56].
Carnitine is used as a biochemical marker of mitochondrial dysfunction. Low levels of total or free carnitine are markers of several diseases associated with primary mitochondrial dysfunction. Carnitine plays an important role in fatty acid metabolism but also serves another purpose. Carnitine can bind to fatty acids in the blood as acylcarnitines and can also bind to excess organic acids. This increases the amount of carnitine esters and decreases free carnitine. Carnitine can then be excreted in the urine along with these fatty acids or organic acids if in excess, resulting in a loss of carnitine in the body, reducing the total amount of carnitine. Short and medium-chain fatty acids can directly enter the mitochondria, while long-chain fatty acids are bound to carnitine and transported through the carnitine shuttle. Fatty acids are metabolized by β-oxidation, which is a cycle that shortens a fatty acid by two carbons with each turn, producing acetyl-CoA, which directly enters the first step of the tricarboxylic acid cycle, and FADH 2, which directly contributes to the electron transport chain as a substrate for complex II. Elevated levels of Acyl-CoA are markers of diseases associated with primary mitochondrial dysfunction (Figure 2) [57,58,59,60,61].
2.2. Mitochondrial Antioxidant System
In functionally complete mitochondria, the action of the mitochondrial antioxidant system, including glutathione, thioredoxin-2, glutathione peroxidase, phospholipid-hydroperoxide-glutathione peroxidase, and Mn-superoxide dismutase, prevents damage to mitochondrial structures by reactive oxygen species [62,63].
GSH is the most important antioxidant in the cell, and the ratio of GSH to GSSH is the most important biomarker of the redox state of the cell. GSH cannot be produced within mitochondria, so it must be imported. A pathway that produces more GSH requires ATP, an energy molecule produced by the mitochondria, so poor mitochondrial function will result in decreased GSH production and reduced ability of the mitochondria to produce cellular energy. ROS is controlled at the inner mitochondrial membrane by leakage of protons back across the membrane, a process that makes mitochondria less efficient This is accomplished by several proton channels such as uncoupling protein (UCP) [64,65,66,67].
We found that the introduction of chloro-2,4- dinitrobenzene (CDNB,1 mM) into the suspension of mitochondria isolated from the brain of white rats leads to a 62% decrease in the amount of GSH and more than 47% inhibition of Mn-SOD activity. A decrease in mitochondrial membrane potential and an increase in mitochondrial pore opening rate were also detected. Deprivation of the mitochondrial glutathione system leads to a decrease in other antioxidant systems as well, and shapes the development of mitochondrial dysfunction. GSH deficiency in mitochondria leads to increased formation of ROS and nitrogen and oxidation of cysteine-dependent sites of proteins forming the mitochondrial pore. Excess of active nitrogen forms (peroxynitrite, nitrosonium ion) formed at GSH deficiency in mitochondria leads to oxidative modification of Mn-SOD and reduction of its activity. Reduction of Mn-SOD activity promotes a secondary “burst” of free-radical reactions and intensification of oxidative destruction of Red-Oxi - sensitive sites of mitochondrial membrane and formation of persistent mitochondrial dysfunction [68].
2.3. The Production of ROS by Mitochondria and the Lack of Mitochondria
It is believed that a key event in the development of mitochondrial dysfunction after hypoxia/reoxygenation is an increase in the production of reactive oxygen species (ROS) by mitochondria. The main causes of mitochondrial dysfunction include electron overload in the respiratory chain under hypoxic conditions, as well as a decrease in the activity of cytochrome c oxidase and Mn-superoxide dismutase (Mn-SOD). Inhibition of cytochrome c oxidase during subsequent reperfusion contributes to a disruption in the process of electron transfer to the final acceptor in the respiratory chain—the oxygen—which leads to an increase in the production of superoxide radicals by the respiratory complexes [69].
Superoxide is formed in the so-called “parasitic” reactions in the initial part of the mitochondrial respiratory chain (CoQH2 - NAD+ ) with the participation of NADH - CoQH2 - reductase, the activity of which is increased by blockade of cytochrome-C-dependent receptor on the outer surface of the mitochondrial membrane against the background of increased reduced flavins. Besides superoxide, the key role in the development of mitochondrial disorders and the development of apoptosis/necrosis belongs to NO and its more aggressive form – peroxynitrite [70,71].
Neuronal mitochondria are an important source of NO. The presence of a constitutive form of NOS localized in the inner membrane and NO production in mitochondria of hippocampal neurons has been shown. Mitochondrial NOS at suboptimal concentrations of L-arginine is able to produce superoxide. Mitochondrial NOS (mtNOS) is significantly activated in response to the development of glutamate “excitotoxicity” and calcium uptake by mitochondria [72,73].
It should be noted that IL-1b and TNF-a play a certain role in mtNOS activation. mtNOS through the production of a dosed level of NO is able to regulate mitochondrial respiration in norm and at the initial, compensated stages of ischemia, modulating the activity of cytochrome-C-oxidase, complexes I and II of the electron-transport chain and the level of NADPH, FAD and coenzyme Q10, as well as changing the availability of O2 for electron acceptance. Further, the role of mtNOS changes to the cardinally opposite one - it participates in activation of “parasitic” reactions of ROS formation by mitochondria. The thiol-disulfide system deserves special attention in expanding the understanding of the mechanisms of NO cytotoxicity and neuronal death [74,75,76].
Intermediates of the thiol-disulfide system have transport properties with respect to NO, thereby increasing its bioavailability; moreover, many thiols such as glutathione, cystenine, and methionine can significantly limit the cytotoxicity of NO and its derivatives, increasing the chance of neuronal mitochondria to survive ischemia [26,77,78].
2.4. Interaction of ROS and NO with the Mitochondria
When electron transfer between the components of the respiratory chain is disrupted, the generation of superoxide anion (O•–2) by mitochondria is significantly enhanced. A deficiency in the functions of the mitochondrial antioxidant system will contribute to the development of oxidative stress, activation of self-sustaining processes of lipid peroxidation, and oxidative damage to proteins and nucleic acids within the mitochondria [79].
The increase of NO concentration in mitochondria observed in the post-ischemic period leads to the interaction of NO with heme iron and paired thiol groups, forming dinitrosol iron complex (DNIC). DNIC, in contrast to NO, is a stronger nitrosylating agent, interacting with protein thiols, histidine, aspartate, glutamine, methionine, cysteine, glutathione and forming N- and S-nitrosothiols [80].
Cerebral ischemia is accompanied by a sharp shift of thiol-disulfide equilibrium towards oxidized thiols, a drop in the activity of enzymes of the thiol-disulfide system (glutathione reductase, glutathione-S-transferase). When glutathione-dependent enzymes are inhibited under ischemia conditions, even greater oxidative modification of low molecular weight thiols occurs, homocysteine formation and, as a consequence, NO transport is impaired with the formation of its cytotoxic derivatives that further enhance thiol oxidation. The presence in a neuron of a sufficiently active thiol antioxidant system capable of regulating NO transport provides cell resistance to nitrosative stress, the earliest neurodegenerative mechanism under ischemia conditions [81,82,83]. It is known that in the first minutes of brain ischemia, NO (macrophagal or exogenous) inhibits oxidative phosphorylation in mitochondria of target cells due to reversible binding to mitochondrial cytochrome C-oxidase. Suppression of electron transport in mitochondria, as shown above, leads to generation of superoxide and, as a consequence, to the formation of ONOO- [26].
Excess superoxydradical, peroxynitrite further oxidizes the thiol groups of the cysteine-dependent site of the mitochondrial inner membrane protein (ATP/ADP-antiporter), which turns it into a permeable nonspecific channel pore. The mitochondrial pore is a channel that passes through both mitochondrial membranes and consists of three proteins: an adenine nucleotide translocator, a potential-dependent anion channel (porin), and a regulatory benzodiazepine receptor. The anti-apoptotic bcl-2 and pro-apoptotic Bax proteins are associated with the benzodiazepine receptor. These proteins are components of the outer membrane and cytosol and participate in apoptosis by controlling the release of cytochrome C [84,85].
Under conditions of oxidative stress, the mitochondrial pore binds to Ca2+ and substances with large molecular weight can pass through the membrane pore. This leads to a drop in membrane potential and matrix swelling, the integrity of the outer membrane is inevitably compromised, and apoptosis proteins are released from the intermembrane space into the cytoplasm [86].
There are several pro-apoptotic proteins: APOptosis-inducing factor (AIF), second mitochondria-derived activator of caspases (Smac) and some procaspases. The inducing factor goes directly to the nucleus, where it causes DNA degradation. Along with specific apoptosis proteins, cytochrome C, which normally serves as the final link in the electrotransport chain, leaves the mitochondrion through the open pore. In the cytoplasm, this protein binds to the protein Apaf-1 (APOptotic protease activating factor-1) and forms an apoptosome complex. With the help of Smac and another factor (Omi/HtrA2), it activates procaspase-9, which, becoming caspase-9, transforms two other proenzymes into caspases-3 and -7; and they already cleave structural proteins, leading to the appearance of biochemical and morphological signs of apoptosis in the neuronal cell [87,88].
2.5. Disorder in the Lipid Layer of Mitochondrial Membranes
In parallel with the above-mentioned destructive processes in the mitochondrial matrix, oxidative changes in the lipid layer of mitochondrial membranes are observed: the level of phospholipids decreases and free fatty acids and lysophosphatides accumulate. Depletion and oxidative modification of mitochondrial pool of membrane phospholipids, promotes mobilization of cytochrome C in the intermembrane space, which significantly facilitates its release into the cytoplasm after opening of mitochondrial pores [89,90].
It was found that changes in the lipid composition of mitochondrial membranes lead to impaired functioning of mitochondrial membrane enzymes. The increase in the content of free fatty acids in the inner mitochondrial membrane promotes dissociation of oxidation and phosphorylation processes, which leads to suppression of ATP synthesis [91,92].
ROS are produced through metabolic processes, particularly by Complexes I and III of the electron transport chain (ETC). ROS can be destructive to many vulnerable parts of the mitochondria. Lipid membranes are especially vulnerable to ROS. The integrity of lipid membranes is crucial for key mitochondrial structures, such as the ETC. If the lipid membrane is damaged, protons may leak through the inner mitochondrial membrane, which can reduce the proton gradient responsible for driving Complex V of the ETC to produce ATP. This will decrease ATP production and lower the inner mitochondrial membrane potential [42,93,[42,93,].
3. Genetic and Cellular Factors Involved in Mitochondrial Dysfunction
3.1. Mitochondrial DNA Damage in Mitochondrial Dysfunction
Another mechanism of mitochondrial dysfunction is the accumulation of damage in the mitochondrial genome and the depletion of the mitochondrial DNA pool. Reactive oxygen species (ROS) can also damage mitochondrial DNA (mtDNA), as mtDNA is not protected like nuclear DNA, and mitochondria are inefficient in repairing mtDNA. This can lead to harmful mutations in mtDNA if the damage occurs in a critical genetic region of the mtDNA. Ultimately, these events lead to a decrease in cell function and the accumulation of mutations in both mitochondrial and nuclear DNA. In turn, damage to mitochondrial genes contributes to the disruption of the electron transfer process in the respiratory chain, resulting in further increased production of free radicals in the mitochondria [94,95].
ROS, interacting with nucleic acids, modify bases, deoxyriboses, and also form new covalent bonds. The most significant modification occurs at the bases [96]. When ROS and free radicals interact with thymine, 5,6-dihydroxy-5,6-dihydrothymine isomers are formed. Cytosine, when interacting with ROS and hydroperoxides, forms hydroxylated cytosine. The interaction of ROS with purines leads to the breakage of the imidazole ring of the molecule fragment, resulting in the formation of formamidopyrimidine residues [97].
Among the products of oxidative modification of purines, the most notable are 8-oxoguanine (8-OG) and its tautomer 8-hydroxyguanine (8-OHG). The formation of these products occurs continuously, but it significantly increases under various pathological conditions and can be considered a marker of oxidative stress. Currently, the determination of 8-OHG is a popular, non-invasive method for assessing oxidative stress levels. Under the influence of ROS, deamination of guanine and adenine may occur, leading to the formation of xanthine and hypoxanthine in the DNA molecule, which have mutagenic effects. The consequences of various types of oxidative damage are not the same. For example, thymine glycols and formamidopyrimidines block replication, are cytotoxic, but their mutagenic potential is limited. The most mutagenic is 8-OHG, and if it is present in the template, all replicative DNA polymerases insert dAMP opposite it and carry out a guanine-cytosine to thymine-adenine substitution. In addition, ROS can directly induce DNA strand breaks by cleaving the sugar-phosphate base [98,99]. DNA aberration induces an enhanced stress response in the mitochondria, which ultimately leads to the formation of defective mitochondrial proteins. ROS are capable of causing indirect damage to nucleic acids. ROS trigger the release of calcium from the mitochondria, which subsequently leads to an increase in nuclease activity. Nitric oxide (NO) plays a significant role in DNA damage by causing the deamination of nucleic acids, followed by the activation of NAD-dependent polymerase (PARP), which catalyzes the attachment of ADP-ribose to histone proteins and DNA [100,101].
Additionally, it should be noted that transporting 1 mole of ADP-ribose requires 1 mole of NAD+ and 4 moles of ATP, which, with significant mobilization of PARP following extensive DNA damage, rapidly depletes the cell's energy reserves. ROS also stimulate ADP-dependent ribosylation of glyceraldehyde-3-phosphate dehydrogenase, leading to the subsequent inactivation of the enzyme and disruption of glycolysis reactions. The impairment of the mitochondria's energy-producing function is also associated with the dysfunction of the respiratory chain, caused by mutations in mitochondrial DNA (mtDNA). Such disruptions affect various biochemical functions of the mitochondria, such as the mitochondrial membrane potential, ATP synthesis, the ATP/ADP ratio (which indicates the state of the oxidative phosphorylation system), ROS generation, and mitochondrial turnover. It has been established that once the proportion of ROS-mediated mtDNA deletions exceeds a sensitivity threshold, the mitochondrial membrane potential, ATP synthesis rate, and ATP/ADP ratio sharply decrease [102,103,104].
The main consequences of the aforementioned processes are the disruption of the respiratory chain function, weakening of the mitochondrial antioxidant defense, accumulation of cytotoxic oxidatively damaged proteins and nucleic acids; mobilization of cytochrome C in the intermembrane space, which, after reoxygenation, transforms the mitochondria into a source of free radicals, reduces their ability to synthesize ATP, and increases the mitochondria's sensitivity to thanatogenic signals [105,106]
3.2. Ca2+ and Mitochondrial Dysfunction
A significant impact on the mitochondria is exerted by the increase in the cytoplasmic level of calcium ions due to the disruption of the cell's ionic homeostasis. It has been shown that the increase in calcium ion concentration in the cytoplasm promotes the induction of the release of thanatogenic factors from the mitochondria, initiates lipolysis processes in the mitochondrial membranes, and disrupts the function of respiratory complexes. It has been established that Ca2+ ions activate proteins that facilitate the formation of mitochondrial channels: Bax and Bid, by activating calpains, as well as Bad and Bik, through the activation of calcineurin [107,108,109].
Movement of excessive amounts of calcium ions into the mitochondrial matrix of Ca2+ ions can lead to additional opening of mitochondrial pores. Another of the known mechanisms of the effect of excessive concentration of Ca2+ ions on mitochondrial structures is damage to the mitochondrial membrane due to activation of phospholipase A2 [107,110]. The possibility of stimulation of oxidative stress in the cell by Ca2+ ions through activation of calcium-sensitive isoform of NOS has been shown [111]. It is known that uncontrolled increase in the concentration of neurotransmitters such as glutamate and dopamine in the extracellular space contributes to the development of neurotoxic processes in the affected area of the brain during stroke. It has been established that excessive glutamate induces unregulated Ca2+ ion influx into the cytosol, further impairing the functional activity of mitochondria [112]. Glutamate is the main excitatory neurotransmitter of the CNS, is involved in cognitive functions, along with acetylcholine maintains the level of wakefulness, but in high concentrations is a neurotoxin [113].
Glutamate realizes its effects through a group of ionotropic membrane receptor channels: NMDA, AMPA, kainate receptors. Excitation of glutamate NMDA receptors, which regulate the content of K+, Na+, Ca++, Cl- in the extra- and intracellular space, activates Ca++-channels, which leads to an increase in the flow of extracellular Ca++ into the cell and release of intracellular Ca++ from the depot, activating various enzyme systems [114]. This leads to impaired phosphorylation of proteins, cleavage of phospholipids and release of arachidonic acid, formation of toxic products, free radicals that have cytotoxic, immunogenic and mutagenic effects, damaging cellular DNA and mRNA [115].
Along with the swelling of mitochondria caused by the influx of calcium, the process shifts into the cytoplasm of the cell and extends to the intercellular level, making the hypoxia tissue-wide. This stage of the ischemic cascade can no longer be reversed by restoring oxygen supply or reperfusion, as the deeply damaged mitochondria stop utilizing oxygen and substrates. They combine with sodium and calcium in the cytoplasm to form endogenous soaps, which literally dissolve (wash away) lipid membranes [116,117,118,119].
3.3. Disruption of Native Protein Structure (Unfolded Protein Response – UPR) and Mitochondrial Dysfunction
An additional factor contributing to mitochondrial damage is the "unfolded protein response" (UPR), which is activated under hypoxic conditions. It has now been shown that the endoplasmic reticulum (ER), when under stress mediated by UPR, can promote the development of degenerative changes in mitochondria by releasing calcium ions into the cytoplasm from the ER, along with the membrane-associated ER protein inositol-requiring enzyme 1 (IRE1). The ER stress sensor IRE1 interacts with STIM1, facilitating the influx of Ca++ and initiating calcium-dependent responses, including the activation of NOS-regulated proteins [120,121,122].
UPR normally mediates cell death by activation of the intrinsic apoptotic pathway, but recent studies have shown that in mitochondrial dysfunction there is a strong activation of UPR, which may lead to activation of programmed necrosis pathways such as necroptosis [123]. To date, it is known that mitochondrial dysfunction is possible through inactivation of the hypoxia-inducible transcription factor HIF-1 [124].
Recent studies have established that adaptation to hypoxia at the cellular and subcellular levels is closely associated with the transcriptional expression of late-acting hypoxia-inducible genes, which are involved in regulating multiple cellular and systemic functions and are necessary for the formation of adaptive traits. It is known that at low oxygen concentrations, this process is primarily controlled by the specific transcription factor HIF-1, which is induced by hypoxia in all tissues. Disruption of energy metabolism due to ischemia or hypoxia, and a decrease in the concentration of mitochondrial ATP, leads to the activation of mitochondrial UPR (UPRmt). In the early stages of ischemia, UPRmt "attempts" to restore mitochondrial energy homeostasis, and if unsuccessful, it suppresses the expression of protective proteins, including HIF-1, and initiates cell death mechanisms [125,126,127]. The use of novel specific UPR inhibitors as remedies for mitochondrial dysfunction is of interest.
3.4. HIF-1 and Mitochondrial Dysfunction
Hypoxia-induced factor, discovered in the early 90s, functions as a master regulator of oxygen homeostasis and is the mechanism by which the organism, responding to tissue hypoxia, controls the expression of proteins responsible for the mechanism of oxygen delivery to the cell, i.e. regulates the adaptive responses of the cell to changes in tissue oxygenation [128,129,130].
Currently, more than 60 direct target genes have been identified for it. All of them contribute to the improvement of oxygen delivery (erythropoiesis, angiogenesis), metabolic adaptation (glucose transport, enhanced glycolytic ATP production, ion transport) and cell proliferation. HIF-1 regulated products act at different functional levels. The end result of such activation is an increase in O2 supply to the cell [131,132].
The identification and cloning of HIF-1 have revealed that it is a heterodimeric redox-sensitive protein composed of two subunits: the inducibly expressed oxygen-sensitive subunit HIF-1α and the constitutively expressed subunit HIF-1β (aryl hydrocarbon receptor nuclear translocator – ARNT). By heterodimerizing with the aryl hydrocarbon receptor (AHR), it forms a functional dioxin receptor. Other proteins of the HIF-1α family are also known, including HIF-2α and HIF-3α. All of them belong to a family of basic proteins, each containing a basic helix-loop-helix (bHLH) domain at the amino-terminal end of each subunit, which is characteristic of various transcription factors and essential for dimerization and DNA binding [133,134,135].
HIF-1α consists of 826 amino acid residues (120 kD) and contains two transcriptional domains at the C-terminal end. Under normoxic conditions, its synthesis occurs at a low rate and its content is minimal because it undergoes rapid ubiquitination and degradation by proteasomes. This process depends on the interaction of the primary structure of HIF-1α and its specific oxygen dependent degradation domain (ODDD - oxygen dependent domen degradation) with von Hippel Lindau (VHL), a tumor growth suppressor, which acts as a protein ligase, widely distributed in tissues [136,137,138].
The molecular basis for this regulation is the O2 -dependent hydroxylation of its two proline residues P402 and P564, part of the HIF-1α structure, by one of three enzymes collectively known as “prolyl hydroxylase domain (PHD) proteins, or HIF-1α prolyl hydroxylases,” which is required for binding of HIF-1α to the VHL protein. α-ketoglutarate, vitamin C and iron are also obligatory components of the process. Along with this, hydroxylation of an asparagine residue in the C - terminal transactivation domain (C-TAD) occurs, resulting in suppression of HIF-1α transcriptional activity. After hydroxylation of the proline residue in the ODDD and the asparagine residue, HIF-1α binds to the VHL protein, which makes this subunit available for proteasomal degradation [139,140].
Under conditions of severe oxygen deficiency, the oxygen-dependent process of hydroxylation of proline residues, which is characteristic of normoxia, is suppressed. As a result, VHL (von Hippel-Lindau protein) cannot bind to HIF-1α, and its degradation by the proteasome is limited, allowing for its accumulation. In contrast, p300 and CREB binding protein (CBP) can bind to HIF-1α, as this process does not depend on asparaginyl hydroxylation. This enables the activation of HIF-1α, its translocation to the nucleus, dimerization with HIF-1β, leading to conformational changes and the formation of a transcriptionally active complex (HRE), which triggers the activation of a broad range of HIF-1-dependent adaptive processes aimed at enhancing the synthesis of endogenous cytoprotective proteins. Among the most important proteins in this group are the so-called heat shock proteins, such as HSP70 (heat shock proteins) [141,142].
HIF-1 mediates mitochondrial biogenesis, mitophagy, and mitochondrial dynamics to regulate mitochondrial population. HIF-1 activation induced mitochondrial fission in human models of pulmonary arterial hypertension (PAH) by phosphorylation of DRP1 by serine 616 [143].
HIF-1 can alter the intracellular distribution of mitochondria by regulating their mobility. The mitochondrial movement regulator (HUMMR) is activated by HIF-1α, and mitochondrial transport shifts in the anterograde direction for efficient distribution throughout the neuron. Additionally, HUMMR may help preserve mitochondrial content in axons dependent on HIF-1α. HIF-1 regulates mitochondrial morphology, such as size, shape, and structure, which could underlie functional changes or be secondary to functional regulation. In mitochondria with stimulated HIF-1 expression, there was an increase in energy metabolism reactions and regulation of reactive oxygen species (ROS) production by mitochondria. HIF-1 positively regulates the expression of lactate dehydrogenase A (LDHA) and promotes the conversion of pyruvate to lactic acid under hypoxic conditions. HIF-1 serves as a sensor for ROS, limiting excessive mitochondrial ROS production in response to cytokine stimulation. Overexpression of HIF-1α blocks the reduction of mitochondrial membrane potential (ΔΨm) under hypoxia and inhibits mitochondrial mechanisms that initiate apoptosis. [144,145,146]
There is evidence for a relationship between HIF-1α and iron homeostasis , especially at the mitochondrial level, because Fe 2+ is mobilized in the Fenton reaction, which produces hydroxyl radical (OH . ) from H 2 O 2 and lipid alkoxyl radicals from lipid peroxides. Notably, the mitochondrial antioxidant apparatus allows iron homeostasis to be maintained by limiting H 2 O 2 production and converting lipid peroxides to lipid alcohol using SOD and GPX4, respectively [147,148]
3.5. HSP 70 and Mitochondrial Dysfunction
Enhanced expression of genes encoding HSP is triggered not only by heat stress but also by a number of different factors and pathogens. Evolutionarily, HSP70 is classified as a highly conserved protein, indicating that it performs fundamental cellular functions. The native HSP70 molecule is a dimer with the ability to form highly oligomeric complexes with many structures in the cell, as well as cytosolic and mitochondrial proteins, and has at least 8 isoforms, the exact number and concentration of which depends on the cell type and is controlled by the type of stressor. HSP70 proteins belong to a class of cellular proteins referred to as “molecular chaperones” (chaperone - mediator). Chaperone activity refers to the ability of HSP70 to recognize and bind exposed hydrophobic surfaces of native polypeptide chains, denatured and oxidatively damaged polypeptides. HSP70 are the main participants in the process of folding of newly synthesized polypeptide chains [149].
The ability of HSP70 to fold polypeptides in a specific confarmation is used in normal cellular processes to regulate key signaling molecules such as cell cycle kinases, caspases, steroid hormone receptors and vitamin D receptors. HSP70 are also involved in the processes of polypeptide translocation into mitochondria, in restoring the structure of damaged and denatured proteins, and in the formation of oligomeric protein complexes. In addition to chaperone function, HSP70 function as regulators/modulators of protein proteolysis (ubiquitins, redox reactions, synthesis of proinflammatory cytokines, synaptic transmission, and Ca++-dependent K+-channels. In addition, the ability of HSP70 to stabilize under ischemia conditions the factor HIF 1 was investigated. Thus, under normoxia conditions HSP 70 is in complex with HIF 1 [150,151].
Under hypoxia conditions, HSP70 is displaced from the complex with HIF 1 by ARNT protein, with further exercise of its chaperone function against HIF 1, in addition, these proteins exert unidirectional action with respect to cell protection against oxidative stress during ischemia [149,152,153].
Recent studies have established the neuroprotective activity of HSP70 and HIF 1 aimed at reducing the phenomena of mitochondrial dysfunction and related oxidative stress. We found that HSP70 and HIF 1 in brain ischemia increase the activity of mitochondrial antioxidant enzymes protecting them from oxidative destruction, restore thiol-disulfide equilibrium, normalize the processes of energy metabolism due to folding of respiratory chain proteins, as well as increase the functional activity of mitochondria, eliminating damaged and denatured proteins. Start and regulate the activity of the compesatorial malate-aspartate shuttle mechanism of energy production [153,154].
This statement is confirmed by the works of some authors. The role of increased expression of HSP70 in brain cells (astrocytes) in protecting them from death caused by oxygen starvation has been shown [155,156].
In addition, the ability of a purified HSP70 preparation to enhance the survival of neurons involved in glutamatergic synaptic transmission in the olfactory cortex of rat brain was demonstrated against the damaging effects of severe anoxia [157,158]. Nevertheless, the mechanism of the protective effect of HSP70 is still unclear. Taking into account the data on the ability of HSP70 to enhance neuronal cell viability under hypoxia conditions and the fact of interaction between HSP70 and HIF-1, which plays a primary role in the cellular response to hypoxia, we can assume that HSP70 is involved in the regulation of signaling pathways of the cell response to hypoxic stress at the level of regulation of HIF-1 stability [149].
In addition, the neuroprotective effect of HSP 70 in ischemia is also explained by its antiapoptotic and “mitoprotective” action. Currently, three main pathways of HSP 70 influence on apoptosis processes are postulated in the literature. Firstly, HSP 70 may affect the functioning and signaling of the FasApo1 receptor inside the cell; secondly, HSP 70 may in one way or another affect the release of cytochrome C from mitochondria and, finally, thirdly, HSP 70 may affect the formation of apoptosomes and the activation of the caspase cascade. HSP 70 blocks apoptosis induced by activation of the Fas/Apo1 receptor. After binding to the ligand, the receptor interacts with adaptor proteins, one of which may be the FADD protein [159,160,161,162]
This adaptor protein FADD binds inactive procaspase 8 and promotes its activation upon receptor-ligand binding. Caspase 8 activates caspases 3, 6, and 7 and thereby initiates proteolysis of target proteins, ultimately leading to apoptosis. In addition, caspase 8 can activate the Bid protein, which induces the release of cytochrome c from mitochondria [163,164]
The site of action of HSP 70 in this complex chain of reactions has not yet been precisely determined. An alternative pathway for triggering apoptosis via Fas/Apo1 involves the Daxx protein. The mechanism of action of this protein is not well understood. Normally, Daxx is localized in the nucleus, where it is bound to certain proteins, but it can move to the cytoplasm and play the role of an adaptor protein responsible for triggering the cascade of JNK-kinases by activating Fas/Apo [165,166]
It is assumed that HSP70 is able to move to the nucleus, where it interacts with Daxx, preventing its release into the cytoplasm and activation of the receptor. Previously, it was noted that HSP70 may participate in the regulation of apoptosis not only at the level of the receptor Fas/Apo1, but also at the level of certain intracellular target proteins [167,168]
Indeed, HSP70 has been shown to prevent mitochondria-initiated apoptosis, and different mechanisms of action of heat shock proteins are possible. The drop in membrane potential induced by cerebral ischemia is known to result in the release of cytochrome c from mitochondria. In the cytoplasm, cytochrome C binds to Apaf1 protein, deoxy ATP and procaspase 9, forming the so-called apoptosome [169,170]
Apoptosome formation is accompanied by autocatalytic activation of procaspase 9 and its transfer into the active form of caspase 9. This enzyme activates procaspase 3 and the following caspases involved in the process of apoptosis. HSP70 inhibits apoptosis in the step between cytochrome c release and cleavage of procaspase 9 in the apoptosome. Recently, the literature has provided evidence that HSP70 is able to interact with cytochrome C [171,172]. The question of what part of cytochrome C released from mitochondria binds to HSP 70 remains open. A number of studies have shown that HSP70 binds only a very small fraction of cytochrome C released from mitochondria and, therefore, cannot play a significant role in apoptosome formation [173,174]. HSP 70 is known to prevent the decrease in membrane potential induced by Bax protein, but does not interact with this protein. There is a hypothesis that in the mitochondrial pathway of apoptosis HSP 70 acts at earlier stages of this complex process and prevents disruption of the actin filament structure [154].
Our works established that modeling of chronic cerebral circulatory disturbance by disruption of cerebral blood circulation led to persistent neurological disorders in surviving animals by the 18th day of the experiment, as well as to the disruption of mitochondria ultrastructure of hippocampal CA1 neurons , which was characterized by an increase in the absolute number of damaged mitochondria, more than 11 times in relation to the intact group of animals, as well as a decrease in intramitochondrial HSP70 3,4 in comparison with similar indicators of the group of falsely operated animals [25].
Thus, summarizing the above, we can conclude that HSP 70 and Hif1b proteins are inevitable companions of pathobiochemical reactions that develop during ischemic brain damage and perform a protective function under these conditions, which is realized by means of enhancing the synthesis of antioxidant enzymes, stabilization of oxidatively damaged macromolecules, direct antiapoptotic and mitoprotective action. Such a role of these proteins in cellular reactions during ischemia raises the question of the development of new neuroprotective agents capable of modulating the expression and synthesis of HSP- and HIF-proteins [6].
4. Mitochondrial Dysfunction and Apoptosis
The activation of neuroapoptosis, according to many researchers, is the primary cause of persistent cognitive and mnemonic dysfunctions in the central nervous system (CNS). Neuroapoptosis develops as a cascade process accompanied by the activation (induction of formation) of specific pro- or anti-apoptotic proteins, as well as specialized proteolytic enzymes—caspases. Among the factors triggering apoptosis, the formation of reactive oxygen species (ROS) during the "distorted" pathway of oxidative metabolism in the cell should be noted. Convincing evidence exists that mitochondria play a central role in ROS production and the subsequent development of apoptosis and necrosis. This involves changes in the permeability of mitochondrial membranes due to the formation of specific mitochondrial pore complexes and the initiation of mitoptosis (Figure 3) [175,176].
Under the influence of hydroxyl radicals, mitochondrial pores open, leading to the expression and release of pro-apoptotic proteins into the cytosol. The opening of these pores transforms mitochondria from "power plants" into "furnaces" for oxidation substrates without ATP production. Precise biochemical studies have established that disruptions in tissue oxygenation, hyperproduction of excitotoxic amino acids, a decrease in "normal" Ca²⁺ accumulation by mitochondria, and oxidative damage to mitochondrial membranes by reactive oxygen species (ROS) exacerbate pore opening and the release of apoptogenic proteins from damaged mitochondria.
In this context, the role of one neurotrophic factor—tumor necrosis factor-α (TNF-α)—is significant. TNF-α is associated with the opening of mitochondrial pores, subsequent membrane damage, and the progression of mitoptosis. The mitochondrial pore is a channel traversing both mitochondrial membranes and comprises three proteins: the adenine nucleotide translocator, the voltage-dependent anion channel (porin), and the benzodiazepine receptor. When this complex binds to Ca²⁺, substances with small molecular masses can pass through the membrane pore. This process leads to a decrease in membrane potential and matrix swelling, which inevitably compromises the integrity of the outer membrane. Consequently, apoptosis-related proteins are released from the intermembrane space into the cytoplasm [177,178].
There are several of them: APOptosis-inducing factor, secondary mitochondria-derived activator of caspases (Smac) and some procaspases. The inducing factor goes directly to the nucleus, where it causes DNA degradation. Along with specific apoptosis proteins, cytochrome C, which normally serves as the final link in the electrotransport chain, leaves the mitochondrion through the open pore. In the cytoplasm, this protein binds to the protein Apaf-1 (APOptotic protease activating factor-1) and forms an apoptosome complex. It, with the help of Smac and another factor (Omi/HtrA2), activates procaspase-9, which, becoming caspase-9, transforms two other proenzymes into caspases-3 and 7; and they already cleave structural proteins, leading to the appearance of biochemical and morphological signs of apoptosis [179,180,181]. Among the earliest events, in particular, are the translocation of phosphatidylserine to the outer membrane layer and DNA fragmentation under the influence of ROS and NO [182].
Among the secondary signs, the most characteristic are the "shedding" of the cell from the matrix, membrane wrinkling, nuclear condensation, and the formation of vesicles containing cellular contents—apoptotic bodies [183]. The release of cytochrome c into the cytoplasm is facilitated by a decrease in pH during the development of lactate acidosis and an increase in oxidative modification of mitochondrial proteins and lipids. This latter reaction is directly triggered by ROS, which are inevitably formed as a result of "parasitic" energy reactions [184]. Cytochrome C can be released in response to an increase in Ca²⁺ ion concentration, which triggers pore opening, and its release is also regulated by proteins of the Bcl-2 family [185,186].
They are the ones that regulate apoptosis at the mitochondrial level. Some of the proteins of this large family (Bcl-2, as well as Bcl-xL, Bcl-w, Mcl-1, Al, and Boo) prevent apoptosis; others (Bax, Bad, Bok, Bcl-xS, Bak, Bid, Bik, Bim, Krk, and Mtd) promote its initiation [187,188].
The entire superfamily of Bcl-2-related proteins is considered to be one of the most important classes of apoptosis-regulating gene products. Their ever-expanding list includes both cell death antagonists and apoptosis-inducing proteins. Due to the large number of representatives of this family described to date, they are usually divided into three groups [189,190].
1- Anti-apoptotic proteins Bcl-2, Bcl-xL, Bcl-w, Mcl-1, Bfl-1 and Boo with homology in the BH1, 2, 3 and 4 regions.
2. Pro-apoptotic proteins Bax, Bak, Bad, Bok and Diva with homology in regions BH1, 2 and 3 but not BH4.
3. "BH3-proteins" are pro-apoptotic proteins such as Bik, Bid, Bim, Hrk, and Blk, which share homology exclusively in the BH3 domain.
The combined action of related cell death agonists and antagonists from the Bcl-2 family represents a regulatory switch whose function is determined, at least in part, by selective protein-protein interactions. These proteins are characterized by the ability to form heterodimers in which partners repress each other [191].
Proapoptotic proteins are mostly localized in the cytosol and translocate to the mitochondrial membrane in response to certain stimuli. Some studies claim that Bach proteins translocate from the cytosol to the mitochondria, while others suggest that they undergo conformational changes enhanced by interaction with Bid proteins [192].
Bid proteins are known to be hydrolyzed by caspase-8 and their C-terminal part interacts with mitochondria. Several models have been proposed for the participation of Bcl-2 family proteins in the regulation of protein transfer from the mitochondrion to the cytosol [193,194,195].
1. Since Bax (like other Bcl-2 family proteins) can form pores in the outer mitochondrial membrane, it has been hypothesized that these pores might be large enough to allow cytochrome c to exit. However, this has not yet been confirmed.
2. There is a hypothesis suggesting that Bax interacts with VDAC, resulting in the formation of an even larger channel capable of accommodating cytochrome C. Notably, the conductivity of this channel is blocked by Bcl-xL [196].
3. Bcl-2 can also form channels in the outer mitochondrial membrane that allow adenine nucleotides to pass through. It is hypothesized that Vach closes VDAC, ATP/ADP exchange between mitochondrion and cytoplasm is disrupted, resulting in the opening of RTR. Moreover, all these phenomena are prevented by Bcl 2 [197,198,199].
4. There is also a hypothesis that suggests that the Bcl-2 family protein complex interacts with the giant pore complex in the inner mitochondrial membrane and leads to membrane depolarization, mitochondrial swelling, and cytochrome С release [200,201].
The Bcl-2 protein has a direct inhibitory effect on pore opening, but it does not protect against all permeability change inducers. Because the mitochondrial pore is regulated by a complex that includes Bcl-2 and Bax antagonists, changes in its stoichiometry (e.g., increased Bax synthesis or Bcl-2 modification) may contribute to permeability changes. It is hypothesized that the ratio between Bcl-2 and Bax proteins, and their phosphorylation, promotes either cell survival (excess Bcl-2 or Bcl-xL) or cell death (excess Bax, phosphorylation of Bcl-2). At the level of phosphorylation, there is a link between changes in mitochondrial permeability and receptor signaling pathways, as phosphorylation can be produced by protein kinases such as JNK (activated by various stress stimuli and through TNFR and Fas receptors, phosphorylates Bcl-2) and PKB/Akt, which transmits signals of growth factors NGF, IGF-1, phosphorylates Bad, preventing apoptosis [202,203,204,205,206,207,208].
Bcl-2 acts as a neuroantioxidant - it blocks the output of cytochrome C and prevents the development of apoptosis. Protein 53 (p53) takes part in triggering apoptosis caused by DNA damage, activation of oncogenes and hypoxia by interacting with Vach, stimulating “death receptors” and apoptosis genes. p53 activates the suicide modulator PUMA (p53 upregulated modulator of APOptosis), which then binds Bcl-2 and disables this protein that prevents apoptosis. Thus, the release of cytochrome C from mitochondria is no longer restrained by anything [109,209]
Some calcium ion-binding proteins, such as ALG-2, encoded by the gene of the same name (APOptosis-linked gene-2), also participate in the development of neuroapoptosis. Thus, the interaction between ALG-2 and the Alix protein (ALG-interacting protein X, also known as AIP1) regulates neuroapoptosis [210,211,212].
It is assumed that in addition to the participation of dysfunctional mitochondria in apoptosis processes, they play a key role in the cell's choice of a pathway to realize the type of morphological death. Data from various researchers indicate that activation of the mechanisms of one or another form of cell death pathway may be determined by the number of open pores in dysfunctional mitochondria. If pores are formed in several mitochondria, the autophagy process is activated in the cell [213,214,215,216]. When pores open in more dysfunctional mitochondria, apoptosis is initiated in the cell, which is probably a consequence of an increase in cytochrome C and apoptosis induced factor (AIF) in the cytoplasm. Eventually, when PT pores open in the cell in almost all dysfunctional mitochondria, dissociation of oxidation and phosphorylation and intensive ATP hydrolysis by mitochondrial ATPase occur, the mechanisms of necrosis-like cell death are activated [217,218,219,220]. In dysfunctional mitochondria the minimum number of open pores does not fundamentally affect the process of cell death, and with a larger number of open pores the initiation of apoptosis is possible, with generalized opening of mitochondrial pores the process of necrosis is realized [221,222,223]. The level of ATP production in dysfunctional mitochondria is of great importance in the “choice” between the realization of apoptosis and necrosis-like programmed cell death. At low ATP level in dysfunctional mitochondria the process of programmed cell death by necrosis mechanism proceeds, sufficient energy supply promotes the process of programmed cell death by apoptosis mechanism [224,225,226,227,228].
5. Possible Strategies for Pharmacocorrection of Mitochondrial Dysfunction
It is shown that the development of mitochondrial dysfunction is naturally accompanied by damage to brain matter [229,230,231]. The following ways of possible pharmacologic correction of mitochondrial dysfunction are currently postulated [6,232,233,234,235,236,237].
1) increase in the efficiency of mitochondria use of deficient oxygen due to prevention of dissociation of oxidation and phosphorylation, stabilization of mitochondrial membranes;
2) weakening of inhibition of Krebs cycle reactions, especially by maintaining the activity of succinatoxidase link;
3) compensation of the lost components of the respiratory chain;
4) formation of artificial redox systems that shunt the electron overloaded respiratory chain;
5) economization of oxygen utilization and reduction of oxygen demand of tissues or weakening of respiratory control in mitochondria, or inhibition of pathways of its consumption that are not necessary for emergency maintenance of vital activity in critical states (non-phosphorylating enzymatic oxidation - thermoregulatory, microsomal, etc., non-enzymatic oxidation of lipids);
6) increase in ATP formation during glycolysis without increasing lactate production;
7) reduction of ATP consumption by the cell for processes that do not determine emergency maintenance of vital activity in critical situations (various synthetic reductive reactions, functioning of energy-dependent transport systems, etc.);
8) introduction of high-energy compounds from outside.
Today, there is no drug that would influence all the above-mentioned ways of correction of energy metabolism. At present, in order to correct mitochondrial dysfunction and energy deficiency, mainly antihypoxants are used - agents that improve the body's absorption of oxygen and reduce the brain's need for it, thereby contributing to the body's resistance to oxygen deficiency. From the biochemical point of view, hypoxia is a violation of substrate oxidation in the tissues of the body due to the impediment or block of electron transport in the respiratory chain, so the action of antihypoxants should be realized at the cellular level and be directed at the respiratory chain. To date, there is no single established classification of antihypoxants. This is due to the fact that the drugs are represented by compounds from different chemical classes and the mechanism of their action is not always studied. They can improve the oxygen transport function of blood or preserve the energy status of the cell under hypoxia. Direct energizing action of antihypoxants is aimed at correction of respiratory chain function under hypoxia conditions. In addition, there are antihypoxants of nonspecific action, the effects of which are aimed at the correction of functional-metabolic systems [238,239,240,241].
Currently, the mechanism of action of known antihypoxants is aimed at the following links of cell energy metabolism and includes drugs of different pharmacological groups:
1. Electron carriers in the respiratory chain (coenzyme Q10 and its analog idebenone, succinic acid, vitamins K1, K3);
2. cofactors of enzyme reactions of energy metabolism (nicotinamide, riboflavin, L-carnitine and others);
3. correctors of lactate acidosis (dimefosfon).
Clinical and experimental studies of the last decade have established that in the acute period of ischemic stroke, pharmacological correction of energy supply should include restoration of electron-transport and conjugating function of the NAD-dependent part of the respiratory chain, as well as activation of compensatory metabolic mechanisms providing electron delivery to the cytochrome section of the chain [6,242,243,244]
5.1. Idebeon
Idebeon is an original drug of quinone structure, is a derivative of ubiquinone (coenzyme Q) Idebeon has reduced hydrophobicity, ability to penetrate the blood-brain barrier, activates the respiratory function of mitochondria and has a positive effect on the processes of free-radical oxidation in brain tissue. Under the conditions of in vitro experiment in nervous tissue culture it was shown that idebenone prevents the formation of free radicals in cytosol and mitochondria, while in parallel reducing the concentration of marker products of oxidative modification of protein molecules. Idebenone is able to act as an electron carrier in the respiratory chain of mitochondria, increases the formation of ATP, and also increases glucose utilization in nervous tissue, in parallel, reducing the likelihood of lactate acidosis development [245,246,247]. In clinical trials, idebenone has demonstrated the ability to improve memory and learning ability in ischemic brain damage. The effectiveness of idebenone in multi-infarct dementia was confirmed by the results of multicenter studies. In cerebrovascular insufficiency of various degrees of severity, a course of therapy with idebenone at a dose of 90 mg/kg resulted in some improvement of cognitive functions after 1.5-2 months of treatment. The efficacy of idebenone in complex therapy of such primary mitochondrial dysfunctions as MELAS syndrome, Leber's optic atrophy, Ley's disease has also been shown [248,249,250].
5.2. Menadione
Menadione or 2-methyl-1,4-naphthoquinone can be incorporated into the respiratory chain and by shunting the electron flow at the site from NADH KoQ, restores the electron flow to cytochrome oxidase under hypoxia. Menadione administration to nerve cell cultures under conditions of moderate hypoxia has been shown to normalize NADH/NAD , increase mitochondrial respiration rate, and increase ATP concentration. Menadione Nrf2-dependent activation of gene expression followed by enhanced formation of protective proteins such as enzymes involved in glutathione biosynthesis [251,252,253]. Menadione was considered as a promising mitochondrial antioxidant and mitoprotector [6]. However, higher concentrations of menadione cause toxic oxidative stress associated with tissue damage, mitochondrial DNA damage, and cell death [254]. Menadione can undergo one-electron reduction, resulting in the formation of unstable free radicals that produce reactive oxygen species by rapid reaction with oxygen, thereby causing oxidative stress. However, other studies have shown that menadione is an effective inhibitor of lipid peroxidation in microsomes by suppressing lipid peroxide formation through various mechanisms, including binding to one-electron molecule transfer enzymes. According to other studies, reduced forms of menadione may exhibit antioxidant activity [255]. All of the above requires additional studies.
Synthetic ubiquinone-containing preparations from the class of redox polymers, among which Sodium polydihydroxyphenylene thiosulfonate - olifen (hypoxen) has gained popularity, contribute to the restoration of respiratory chain functioning.
5.3. Olifen
Olifen has a polyphenolic ubiquinone component in its structure, contributes to the reduction of electron leakage from the respiratory chain and exhibits antihypoxic and antioscidatic properties. Under conditions of oxygen deficiency the use of oliphene is accompanied by oxidation of reduced nicotinic and flavinic nucleotides. It prevents the development of lipoperoxidation reactions of mitochondrial suspension membranes, preserves the charge of the outer mitochondrial membrane, stimulates the destruction of peroxidation products [6]. Olifen is used in the manifestation of secondary mitochondrial dysfunction due to chronic myocardial ischemia and working hypoxia, due to physical overload. It shows actoprotective effect [256,257]
5.4. Succinic Acid
To enhance the alternative NADH-oxidase pathway of ATP formation, agents involved in succinatoxidase oxidation are used, the stimulation of which is achieved by activation of succinate dehydrogenase reaction through exposure to succinate-containing compounds that facilitate the transport of succinate into the cell. Succinate exerts its antihypoxic effect in two ways. First, it acts as a substrate of the tricarboxylic acid cycle and the enzyme succinate dehydrogenase. Second, it plays a role as a signaling molecule, activating HIF-1α and the orthologous receptors SUCNR1 and GPR91. Interaction with the latter promotes an increase in the level of reabsorbed glucose and stimulation of gluconeogenesis [258,259,260]. The combination of sodium succinate and cytochrome C is promising from the point of view of energetotropic and anti-ischemic action. Sodium salts of succinate are effective in reducing metabolic intracellular acidosis due to intracellular oxidation with the replacement of one hydrogen molecule by sodium to form bicarbonate. Antioxidant action of succinates is realized due to inhibition of production of reactive oxygen species by bioenergetic reactions of mitochondria. Antioxidant effect of succinate is manifested in the reduction of oxidative stress products, in particular carbonylated proteins in the susceptibility of mitochondria of myocardium and brain of experimental animals. Activates the synthesis of endogenous antioxidant - glutathione. Stimulates erythropoiesis. Increases the level of adrenaline, noradrenaline and dopamine, due to which psychostimulant, normotimic and antidepressant effects are observed. The efficacy of the drug containing succinate has been shown in secondary mitochondrial dysfunctions caused by myocardial ischemia, chronic cerebral ischemia and working hypoxia [261]. The possibility of using succinic acid preparations in primary mitochondrial dysfunction is actively discussed, particularly in a patient with MELAS syndrome [262,263]. Combination succinate-containing drugs such as reamberine, cytoflavin, and remaxol have found use in clinical practice [261,264,265]. The bioavailability of succinate is enhanced by combining it with various metabolites (such as citric and malic acids). Salts of succinic acid and mixtures, such as Limontar (a combination of sodium succinate and citric acid), are more readily accessible to mitochondria and are oxidized within them. A more promising approach today involves increasing the activity of succinate dehydrogenase and stimulating endogenous succinate production. This is achieved through pharmacological agents that act as succinate precursors and improve its penetration through the blood-brain barrier. However, such drugs have not yet found application in clinical medicine, and research in this area is currently limited to experimental studies [6,266,267].
Cytochrome C and KoQ preparations may be recommended in later stages of hypoxia as a redox mediator of the respiratory chain at the site between flavoprotein dehydrogenase and cytochromes stabilizes mitochondrial inner membranes, relieves succinate oxidase and NADH-oxidase inhibition [268,269,270]
In the 1960s–1970s, under the leadership of Academician V. Skulachev, a class of mitochondrial antioxidants was developed under the general name SkQ. To deliver antioxidants to mitochondria, a low-molecular-weight compound was proposed, consisting of a positively charged phosphorus atom surrounded by three hydrophobic phenyl groups (triphenylphosphonium, TPP, a charged triphenylphosphine). The mitochondrial protective properties of compounds combining TPP with ubiquinone and TPP with plastoquinone (a quinone involved in the electron transport chain during photosynthesis) were studied.
Currently, 11 compounds of this type are known: SkQ1, SkQR1, SkQ2, SkQ2M, SkQ3, SkQ4, SkQ5, SkQBerb, SkQPalm, C12TPP, and MitoQ. Plastiquinone derivatives with one, two, or three methyl groups are used as the antioxidant component. SkQR1 is considered the most active.
The mechanism of the mitochondrial protective action of such compounds involves their penetration into mitochondria and the reduction of reactive oxygen species (ROS). This occurs both by decreasing ROS production during mitochondrial bioenergetic reactions and through direct interaction between SkQ compounds and ROS [271,272,273,274,275].
No less important drugs in conditions of cerebral hypoxia will be the so-called “correctors of lactate acidosis”. These medications include dimefosfon.
5.5. Dimefosfon
Dimefosfon is a dimethyl ester of 1,1-dimethyl-3-oxobutyl phosphonic acid. Dimefosfon had pronounced antihypoxic and anti-ischemic properties. The antihypoxic effect of dimefosfon was associated with a decrease in lactate production in brain structures simultaneously with an increase in the activity of key cellular energy supply enzymes: NADPH (diaphorase, succinate dehydrogenase and glycero-6-phosphate dehydrogenase), accompanied by an increase in ATP and creatine phosphate. Dimefosfon increased brain energy charge by decreasing oxygen consumption by brain tissue. Dimefosfon inhibits lipoperoxidation processes of mitochondrial meebras isolated from rat hippocampal neurons. It is active in relation to secondary mitochondrial dysfunction caused by cerebral ischemia. It is known to be a positive modulator of neuronal acetylcholoesterase. [6].
However, despite the accumulated large clinical material on the use of the above-mentioned drugs, at present, they do not fully meet all the requirements for drugs correcting mitochondrial dysfunction. Thus, clear criteria and principles of their combined use, as well as the possibility of their use at different stages of bioenergetic hypoxia have not been developed. In addition, drugs are used chaotically, without sufficient knowledge about their capabilities, as well as without planning the treatment strategy from the position of expediency. In addition, a significant disadvantage of modern antihypoxants and neuroprotectors is their inability to affect the delicate molecular and biochemical chains of energy metabolism at already formed mitochondrial dysfunction, as well as their ineffectiveness with respect to the processes of cell death [6,276,277].
In this regard, another approach to correcting mitochondrial dysfunction is currently being considered—the use of antioxidant drugs that, by acting on the cell's antioxidant system, reduce the concentration of cytotoxic marker products of oxidative destruction of proteins and nucleic acids. Through this effect, antioxidants can restore energy production in the mitochondrial respiratory chain and mitigate mitochondrial dysfunction, given the well-known role of reactive oxygen species (ROS) in its development.
However, it is important to emphasize that the use of antioxidant drugs is often inconsistent, and dosing regimens for these drugs are not clearly established. This can lead to a situation where antioxidants begin to act as pro-oxidants within cells.
In this context, so-called "thiol antioxidants," which contain free SH-groups in their molecular structure, are of particular interest. According to several researchers, the presence of SH-groups allows thiol antioxidants to compete with ROS, forming "stable complexes" with them, thereby protecting the SH-groups in the cysteine-dependent region of mitochondrial inner membrane proteins. This action of antioxidants helps prevent the opening of mitochondrial pores under conditions of oxidative and nitrosative stress [6,278,279].
Recently, the thiol antioxidant N-acetylcysteine (NAC) has attracted the attention of pharmacologists and clinicians.
5.6. NAC (N-Acetylcysteine)
NAC acts as a "trap" for peroxynitrite and nitric oxide (NO), suppresses the production of IL-1β, and inhibits the activity of H₂O₂-dependent p38 stress kinases in astrocytes. It has been established that NAC, indirectly through the reduction of ROS levels, inhibits the functioning of the MAP kinase cascade, thereby decreasing the production of transcription factors. This, in turn, reduces the expression of genes responsible for the synthesis of NO synthase and COX-1 in astrocyte cultures [280,281,282].
It should be noted, however, that studies on the activity of NAC have been conducted predominantly in vitro and on model pathologies associated with brain ischemia, where it demonstrated relatively low therapeutic efficacy. Among other antioxidants, derivatives of hydroxypyridine—emoxypine and its succinate salt, mexidol—have found practical applications.
5.7. Emoxypine and Mexidol
Emoxypine and mexidol are highly effective inhibitors of free radical oxidation and suppress oxidative protein modification in ischemic brain tissue. Both drugs exhibit pronounced membrane-stabilizing effects (increasing the content of phosphatidylserine and phosphatidylinositol) and normalize the activity of various membrane-dependent enzymes, such as adenylate cyclase and creatine phosphokinase [6,283,284].
5.8. Ethylmethylhydroxypyridine Succinate
Ethylmethylhydroxypyridine succinate (Mexidol, Mexicor) is widely used today as an antioxidant and antihypoxant. It activates compensatory metabolic processes by facilitating succinate entry into the mitochondrial respiratory chain, thereby enhancing the energy-producing function of mitochondria, improving energy metabolism, and maintaining the levels of macroergic compounds under conditions of mitochondrial insufficiency.
Mexidol inhibits lipid peroxidation and increases the activity of antioxidant enzymes, such as superoxide dismutase and glutathione peroxidase, due to its 3-hydroxypyridine component. Additionally, Mexidol modulates the activity of membrane-associated enzymes (adenylate cyclase, acetylcholinesterase, etc.), neurotransmitter transport systems, ion channels, receptors, and receptor complexes (such as acetylcholine and GABA).
It also has the ability to reduce glutamate excitotoxicity and nitric oxide (NO) levels, while regulating the expression of early-response genes such as c-fos and HIF-1mRNA [285,286].
Mexidol exerts a protective effect on the protein components of neuronal membranes, including receptors and ion channels, enhancing nerve conduction and synaptic transmission. It stimulates the energy-producing functions of mitochondria and ATP synthesis. Mexidol has been shown to improve the ultrastructure of myocardial mitochondria following ischemia and reperfusion.
In experimental studies, Mexidol alleviated cognitive deficits in animals during the recovery period after total brain ischemia, under conditions of emotional-pain stress, and following the administration of cholinomimetics. Mexidol demonstrated good therapeutic efficacy in the treatment of carotid stroke, promoting regression of neurological and cognitive deficits, normalizing EEG patterns, and showing no significant side effects [6,287].
Mexidol is used mainly in secondary mitochondrial dysfunctions accompanying chronic cerebral ischemia, CNS damage due to prenatal hypoxia, multiple sclerosis, working hypoxia [264,288]. In primary mitochondrial dysfunctions mexidol was used as part of complex therapy of MELAS syndrome and in mitochondrial myopathy [6,289].
5.9. Meldonium
Meldonium (mildronate) is a metabolitotropic cardioprotector. The main mechanism of its action is reversible inhibition of the rate of carnitine synthesis from its precursor - γ-butyrobetaine, which leads to a decrease in carnitine-mediated transport of long-chain fatty acids without changing the metabolic processes of short-chain fatty acids through mitochondrial membranes, i.e. there is no complete blockage of oxidation of all fatty acids [6,290]. Meldonium in secondary mitochondrial dysfunction due to myocardial ischemia, working hypoxia, cardiomyopathy after prenatal hypoxia, cerebral ischemia [291,292,293]. There have been attempts to use meldonium for the treatment of primary mitochondrial dysfunction, particularly MELAS syndrome [6].
The thiol antioxidant and ROS/NO scavenger Thiotriazoline has gained wide international recognition. It was first synthesized in the USSR, specifically at the Zaporizhzhia Medical University in 1982, and became an original pharmaceutical drug in Ukraine in 1992.
Thiotriazoline enhances the energetic potential of the myocardium and mitigates oxidative stress in conditions such as ischemic heart disease, physical overexertion, and work-related hypoxia [6].
5.10. Thiotriazoline
Thiotriazoline (tiazotic acid (thiotriazoline) The results of numerous studies have shown the ability of thiotriazoline to influence oxidative processes in mitochondria in ischemic lesions of the brain and heart (Figure 4). Thus, it was found that the anti-ischemic efficacy of the drug is based on its ability to reduce the degree of inhibition of oxidative processes in the Krebs cycle, activate the compensatory malate-aspartate shuttle mechanism and increase the production of ATP and ADP against the background of a decrease in AMP [294]. Under conditions of ischemic brain injury, Thiotriazoline normalizes glucose utilization within cells, increases the activity of glucose-6-phosphate dehydrogenase, restores the NAD/NADH ratio and cytochrome C oxidase activity, and elevates the levels of pyruvate, malate, isocitrate, and succinate. Simultaneously, it reduces lactate overproduction, mitigates uncompensated acidosis, and counters its pro-oxidant effects. Thiotriazoline is the only drug known to activate the conversion of lactate to pyruvate
In both in vitro and in vivo studies, Thiotriazoline inhibited mitochondrial ROS production. As a nitric oxide (NO) scavenger, it enhances NO bioavailability. Moreover, it prevents the irreversible inactivation of the NF-kappa B transcription factor by protecting cysteine residues—Cys 252, Cys 154, and Cys 61—in its DNA-binding domains from excessive ROS. Thiotriazoline also appears to facilitate the restoration of these residues during reversible inactivation, acting in the role of Redox Factor-1.
Preincubation of mitochondrial suspensions with Thiotriazoline (10⁻⁵ M) and subsequent addition of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (60 μM), sodium nitroprusside (100 μM), or H₂O₂ (50 μM) significantly inhibited the rate of mitochondrial pore opening (p≤0.05), increased mitochondrial membrane potential (p≤0.05), and raised intramitochondrial GSH concentration (p≤0.05).
Thiotriazoline preserves the threshold sensitivity of membrane receptors, maintains membrane fluidity, protects phospholipids from oxidation, prevents ion channel polarization, and normalizes ion transport. Additionally, Thiotriazoline inhibits NO-dependent apoptotic mechanisms and increases levels of the anti-apoptotic [6].
Our studies have established that tiotriazolin is effective in secondary mitochondrial dysfunction due to myocardial ischemia, cerebral ischemia, working hypoxia after exercise, and intrauterine hypoxia. And also congenital mitochondrial dysfunction leading to myocardial hypertrophy in children [149,295,296,297,298,299].
5.11. Angiolin
It has been established that in cases of already developed mitochondrial dysfunction and the initiation of apoptotic processes, metabolic therapy agents (such as coenzyme Q10, carnitine, B vitamins, and succinic acid derivatives) demonstrate limited efficacy and are unable to regulate the delicate aspects of energy metabolism for which they serve as intermediates (Figure 5).
Another approach to correcting mitochondrial dysfunction involves the use of thiol antioxidants. These agents compete with the SH-groups of cysteine-dependent sites on the inner mitochondrial membrane protein (ATP/ADP antiporter) for reactive oxygen species (ROS) and peroxynitrite, forming stable complexes with the latter. This mechanism prevents the opening of mitochondrial pores under conditions of oxidative and nitrosative stress.
This foundation led to the development of a fundamentally new metabolitotropic endothelial protector with an original structure—((S)-2,6-diaminohexanoic acid 3-methyl-1,2,4-triazolyl-5-thioacetate), named "Angiolin". Angiolin exhibits anti-ischemic, cardioprotective, neuroprotective, and antioxidant properties [295,300,301,302].
The mitochondrial protective effect of Angiolin was established. Pre-incubation of the mitochondrial suspension with Angiolin (10-5 M) and the addition of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (60 μM) significantly inhibited the rate of mitochondrial pore opening (p≤0.05) and increased the mitochondrial membrane potential (p≤0.05). Additionally, it elevated the intramitochondrial concentration of HSP70 (p≤0.05) and reduced (p≤0.05) the number of damaged mitochondria compared to control samples [6].
Angiolin (100 mg/kg) is effective in secondary mitochondrial dysfunction following chronic cerebral ischemia and intrauterine hypoxia. Angiolin significantly reduced the number of damaged mitochondria in CA1 hippocampal neurons and increased the intramitochondrial concentrations of HSP70 and GSH [300,303,304].
5.12. Benzodiazepines
Recent studies have shown that the manifestations of mitochondrial dysfunction in CNS ischemic lesions can be reduced by regulating the opening of mitochondrial pores. Given that the mitochondrial pore contains a regulatory benzodiazepine receptor, benzodiazepine receptor modulators deserve attention. Among them, the derivatives of 1,4-benzodiazepine and 1,3,4-benzotriazepine are of the greatest interest, as they have a wide range of neurotropic activities: antidepressant, antihypoxic, and nootropic effects. However, the effect of these drugs on mitochondrial functional activity during brain ischemia has not been studied, which defines the potential for further research in this area [305,306,307,308]. Pre-incubation of the mitochondrial suspension with a benzodiazepine receptor modulator (cinazepam, 10-5 M) and the addition of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (60 μM) showed a weak inhibitory effect on the rate of mitochondrial pore opening, increased the mitochondrial membrane potential, and slightly increased the intramitochondrial concentration of HSP70. However, it did not significantly affect the number of damaged mitochondria compared to the control samples [6].
5.13. Estrogens and Selective Estrogen Receptor Modulators.
It is known that, alongside their effects on the reproductive system, estrogens have a multifaceted impact on higher brain functions. It has been shown that estradiol plays a key role in the prenatal and early postnatal development of the brain, as well as throughout life in various disorders of the hypothalamic-pituitary-ovarian system. The main mechanism of estrogen action is genomic, which occurs at the level of the cell nucleus; the non-genomic mechanism exerts its effect through the cell membrane. To implement genomic effects in the brain, there are two types of receptors – estrogen receptor α (ERα) and estrogen receptor β (ERβ).
Tamoxifen and livial, which are anti-estrogens for breast tissue, exert their anti-estrogenic action through the activation of corepressors. Conversely, tamoxifen and livial, being agonists for endometrial cells, exhibit estrogenic effects through the stimulation of coactivators, while livial has no effect on endometrial cells [309,310,311,312].
Different concentrations of adapter proteins in estrogen-sensitive cells determine the selectivity and agonist/antagonist nature of tamoxifen and livial. It has been established that tamoxifen and its metabolite can inhibit the neurotoxic glutamate system and exert neuroprotective effects in β-amyloid-induced amnesia [313,314]. Experimental studies have established a rather high neuroprotective efficiency of estrogen receptor modulators in conditions of deprivation of the glutathione link of the thiol-disulfide system in the suspension of isolated neurons. Such an effect of estrogen receptor modulators, in our opinion, is explained, first, by their direct antioxidant effects. Secondly, it is known that estrogen receptor activation involves uncoupling of estrogen receptors from heat shock proteins (hsp), which ensures the penetration of HSP70 inside the cell.
Thirdly, it has been established that selective estrogen receptor modulators modulate the expression of global transcription factors, particularly AP-1, which is responsible for the synthesis of key enzymes in the antioxidant and thiol-disulfide systems. This explains the significant ability of tamoxifen and livial to restore the activity of superoxide dismutase, glutathione S-transferase, and glutathione reductase. Additionally, the anti-apoptotic effect of estrogens has been noted, which is due to the stimulation of the expression of anti-apoptotic proteins from the Bcl-xL family. Thus, our results reveal the importance of the neuronal glutathione system as a key target for neuroprotective therapy and provide experimental justification for the clinical use of estrogen receptor modulators—tamoxifen and livial—as neuroprotective agents. Tamoxifen is effective in secondary mitochondrial dysfunctions resulting from cerebral ischemia. Our experimental studies have established the neuroprotective activity of the selective estrogen receptor modulator tamoxifen citrate, which occurs, firstly, due to its ability to increase the levels of Hsp70 proteins in the brain tissue during the acute phase of ischemia. Secondly, tamoxifen citrate is capable of limiting the development of oxidative and nitrosative stresses, leading to a reduction in the concentrations of homocysteine and nitrotyrosine in the brain and an increase in glutathione, thus restoring thiol-disulfide balance in nerve cells. The synergistic enhancement of these effects of tamoxifen citrate under acute cerebral ischemia conditions led to a pronounced neuroprotective effect—reducing mortality and neurological deficit [315,316].
5.14. Neuropeptides
Currently, the search for neuroprotective and energy-tropic drugs of HSP-mediated action is conducted among neuropeptides capable of increasing HIF-protein concentration by modulating the expression of global transcription factors [149].
The discovery of neurotrophic peptide factors, prompted the formation of a new strategy of pharmacotherapy - peptidergic, or neurotrophic therapy of neurodegenerative diseases [317,318]. A new class of neuroprotective agents has been created on the basis of peptide preparations. The ideology of creating these drugs is based on the recognition of the role of neuropeptides as universal “integrators” that unite and coordinate the activities of the three main regulatory systems of the organism - nervous, endocrine and immune systems [319].
A number of drugs with neurotrophic properties have been developed that are successfully used in the therapy of a wide range of neurological disorders. Cerebrolysin, Cerebrocurin, Cortexin, Semax, which have been successfully used in the clinic of neurological disorders for ten years already, have been the most successful (Figure 6) [6,320]. Among the presented drugs, a special place is occupied by the domestic neuropeptide drug – cerebrocurin, which includes amino acids, neuropeptides, as well as low-molecular-weight products of controlled proteolysis of proteins and peptides from cattle embryos. The mechanism of action and targets of cerebrocurin are fundamentally different from other neuropeptide drugs, particularly from cerebrolisin. Cerebrocurin contains peptides that carry a program for analyzing the state and construction of the CNS. Thus, the final effect differs due to the qualitatively different mechanism of action. In addition, cerebrocurin increases the affinity of BDNF for its receptors. When cerebrocurin was administered to animals with cerebral ischemia, an increase in ATP production in oxidative reactions in the brain (cortex, CA1 hippocampus) was observed, as evidenced by an increase in malate content, enhanced activity of mitochondrial malate dehydrogenase, and cytochrome C oxidase. Cerebrocurin not only affected energy production but also its transport and utilization, as indicated by the increase in the activity of mitochondrial and cytoplasmic creatine phosphokinase. An important aspect of cerebrocurin's effect on energy metabolism under brain ischemia conditions was the significant reduction in lactate production and lactate acidosis. Through the regulation of early response gene expression (c-fos) and the anti-apoptotic protein bcl-2, cerebrocurin is able to influence neuroapoptosis processes to some extent in cerebral ischemia conditions. The reduction of mitochondrial dysfunction in the context of ischemic brain pathology and the normalization of neuronal energy metabolism upon cerebrocurin administration contributed to the preservation of the main morphofunctional characteristics of neurons in the sensorimotor cortex during cerebral ischemia. Pre-incubation of the mitochondrial suspension with cerebrocurin and the addition of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahidropyridine) (60 μM) significantly inhibited the rate of mitochondrial pore opening, increased the mitochondrial membrane potential, and elevated the intramitochondrial concentration of HSP70. Moreover, it had a significant effect on the number of damaged mitochondria compared to control samples [6,321].
Experimental studies have revealed that cerebrocurin prevents microglia hyperactivation and reduces the production of IL-1α and other pro-inflammatory cytokines, reflecting the drug's impact on the severity of local inflammatory reactions and processes. Cerebrocurin is effective for pharmacocorrection of both primary mitochondrial dysfunction (MELAS, MERRF) and secondary mitochondrial dysfunction, especially as a result of cerebral ischemia, intrauterine hypoxia, and ischemic opticopathy [322,323,324,325]. These neuropeptide drugs have also been successfully utilized in the management of various comorbid conditions, including hypertension, thyroid pathology, alterations in microbiota, chronic kidney disease, pregnancy, osteoarthritis, and type 2 diabetes mellitus [326,327,328,328,330,331]. Their broad therapeutic potential highlights their applicability across a diverse spectrum of systemic and metabolic disorders [332,333,334,335,336].
6. Conclusions
In summary and synthesizing the literature on the research topic, it can be concluded that antioxidants, selective modulators of estrogen and benzodiazepine receptors, and neuropeptide drugs are increasingly being applied both in model pathologies and in clinical neurology. At the same time, a number of issues related to the neuro- and energetropic effects, mechanisms of action of these drug groups, and especially Thiotriazoline, Cerebrocurin, and the new original drug "Angiolin," require more detailed and in-depth study. All of this underscores the need to investigate the effects of the aforementioned drugs on mitochondrial dysfunction, energy, and energy-dependent processes occurring in brain cells during ischemic injury.
Author Contributions
Conceptualization, I.B., O.P., N.B., V.R., S.P., E.S., V.O. and O.K.; validation I.B., O.P., N.B., V.R..; writing—review and editing, I.B., O.P., N.B., V.R., S.P., E.S., V.O. and O.K.; visualization, I.B., O.P., S.P., E.S., V.O. and O.K.; supervision I.B., O.P., N.B., V.R., S.P., E.S., V.O. and O.K.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cook, A.M.; Jones, G.M.; Hawryluk, G.W.J.; Mailloux, P.; McLaughlin, D.; Papangelou, A.; Samuel, S.; Tokumaru, S.; Venkatasubramanian, C.; Zacko, C.; et al. Guidelines for the Acute Treatment of Cerebral Edema in Neurocritical Care Patients. Neurocritical Care 2020, 32, 647–666. [Google Scholar] [CrossRef] [PubMed]
- Zissimopoulos, J.M.; Tysinger, B.C.; A, St. Clair, P.; Crimmins, E.M. The Impact of Changes in Population Health and Mortality on Future Prevalence of Alzheimer’s Disease and Other Dementias in the United States. Journals Gerontol. Ser. B 2018, 73, S38–S47. [Google Scholar] [CrossRef] [PubMed]
- Chief Medical Officer’s Annual Report 2023 Health in an Ageing Society.
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxidative Med. Cell. Longev. 2020, 2020, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Cherniy, V.I.; Nagornaya, E.A.; Bukhtiyarova, N.V.; Kucherenko, V.I. Neuroprotection and Neuroplasticity. Kiev: Logos, 2015.
- Yang, J.; Guo, Q.; Feng, X.; Liu, Y.; Zhou, Y. Mitochondrial Dysfunction in Cardiovascular Diseases: Potential Targets for Treatment. Front. Cell Dev. Biol. 2022, 10, 841523. [Google Scholar] [CrossRef]
- Weissig, V.; Edeas, M. Targeting Mitochondria 2023 Abstract Book. J. Mitochondria, Plast. Endosymbiosis 2023, 1. [Google Scholar] [CrossRef]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free. Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef]
- Murphy, M.P. Mitochondrial Thiols in Antioxidant Protection and Redox Signaling: Distinct Roles for Glutathionylation and Other Thiol Modifications. Antioxidants Redox Signal. 2012, 16, 476–495. [Google Scholar] [CrossRef]
- Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alsudir, S.A.; Alfahad, A.J.; Alshehri, A.A.; Almughem, F.A.; Mohammed, R.Y.; Alzaydi, M.M. Recent advances in mitochondrial diseases: From molecular insights to therapeutic perspectives. Saudi Pharm. J. 2022, 30, 1065–1078. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: the role of glucose in physiological and pathological brain function. 2013, 36, 587–597. [CrossRef]
- Jain, V.; Langham, M.C.; Wehrli, F.W. MRI Estimation of Global Brain Oxygen Consumption Rate. J Cereb Blood Flow Metab. 2010, 30(9):1598-Epub 2010 Apr Erratum in: J Cereb Blood Flow Metab. 2010, 30(12):Erratum in: J Cereb Blood Flow Metab. 2011, 31(5):1336.
- Raichle, M.E.; Gusnard, D.A. Appraising the Brain's Energy Budget. Proc Natl Acad Sci U S A. 2002, 99(16):10237-9. Epub 2002.
- Watts, M.E.; Pocock, R.; Claudianos, C.; Chowen, J.A.; Arevalo, M.A.; Rosenberger, T.A. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Mei, S.; Wang, X.; Hu, G.; Lu, M. Focusing on mitochondria in the brain: from biology to therapeutics. Transl. Neurodegener. 2024, 13, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Thornton, B.; Cohen, B.; Copeland, W.; Maria, B.L. Mitochondrial Disease: Clinical Aspects, Molecular Mechanisms, Translational Science, and Clinical Frontiers. J Child Neurol. 2014, 29(9):1179-207.
- Johri, A.; Beal, M.F. Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Pharmacol. Exp. Ther. 2012, 342, 619–630. [Google Scholar] [CrossRef]
- Vincent, A.E.; Ng, Y.S.; White, K.; Davey, T.; Mannella, C.; Falkous, G.; Feeney, C.; Schaefer, A.M.; McFarland, R.; Gorman, G.S.; et al. The Spectrum of Mitochondrial Ultrastructural Defects in Mitochondrial Myopathy. Sci. Rep. 2016, 6, 30610. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Mazur, I.A.; Kucherenko, L.I.; Nagornaya, E.A.; Gorbacheva, S.V.; Bidnenko, A.S. The molecular and ultrastructural aspects of the formation of mitochondrial dysfunction in the modeling of chronic cerebral ischemia: The mitoprotective effects of Angiolin. Neurochem. J. 2016, 10, 131–136. [Google Scholar] [CrossRef]
- Belenichev, I.; Popazova, O.; Bukhtiyarova, N.; Savchenko, D.; Oksenych, V.; Kamyshnyi, O. Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection. Antioxidants 2024, 13, 504. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Averina, O.A.; Andreev-Andrievskiy, A.A.; Zinovkin, R.A.; Lyamzaev, K.G.; Marey, M.V.; Egorov, M.V.; Frolova, O.J.; et al. Mitochondrion-targeted antioxidant SkQ1 prevents rapid animal death caused by highly diverse shocks. Sci. Rep. 2023, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Di Lisa, F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J Mol Cell Cardiol. 2015, Jan;78:100-6.
- Krasnikov, B.F.; Zorov, D.B.; Antonenko, Y.N.; Zaspa, A.A.; Kulikov, I.V.; Kristal, B.S.; Cooper, A.J.; Brown, A.M. Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial permeability transition. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2005, 1708, 375–392. [Google Scholar] [CrossRef] [PubMed]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Gutiérrez-Aguilar, M. The still uncertain identity of the channel-forming unit(s) of the mitochondrial permeability transition pore. Cell Calcium 2018, 73, 121–130. [Google Scholar] [CrossRef]
- Park, W.; Wei, S.; Kim, B.-S.; Bae, S.-J.; Chae, Y.C.; Ryu, D.; Ha, K.-T. Diversity and complexity of cell death: a historical review. Exp. Mol. Med. 2023, 55, 1573–1594. [Google Scholar] [CrossRef]
- Orsucci, D.; Ienco, E.C.; Rossi, A.; Siciliano, G.; Mancuso, M. Mitochondrial Syndromes Revisited. J. Clin. Med. 2021, 10, 1249. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.A. Brain Mitochondrial Bioenergetics in Genetic Neurodevelopmental Disorders: Focus on Down, Rett and Fragile X Syndromes. Int. J. Mol. Sci. 2023, 24, 12488. [Google Scholar] [CrossRef]
- Rahman, M.H.; Suk, K. Mitochondrial Dynamics and Bioenergetic Alteration During Inflammatory Activation of Astrocytes. Front. Aging Neurosci. 2020, 12:614410.
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef]
- Goel, P.; Chakrabarti, S.; Goel, K.; Bhutani, K.; Chopra, T.; Bali, S. Neuronal cell death mechanisms in Alzheimer’s disease: An insight. Front. Mol. Neurosci. 2022, 15, 937133. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Makyeyeva, L.; Morozova, O.; Oksenych, V.; Kamyshnyi, O. Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs. Int. J. Mol. Sci. 2024, 25, 10475. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative Stress, Mitochondrial Damage and Neurodegenerative Diseases. Neural Regen Res. 2013, 8(21):2003-14.
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M.; Tsimpiktsioglou, A.; Stampouloglou, P.K.; Oikonomou, E.; Mourouzis, K.; et al. Mitochondria and cardiovascular diseases—from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256–256. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Faller, D.V. Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl Oncogenomics. 2008, 3:53-65.
- Xu, H.; Liu, Y.Y.; Li, L.S.; Liu, Y.S. Sirtuins at the Crossroads Between Mitochondrial Quality Control and Neurodegenerative Diseases: Structure, Regulation, Modifications, and Modulators. Aging Dis. 2023, 14(3):794-824.
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Khoury, N.; Koronowski, K.B.; Young, J.I.; Perez-Pinzon, M.A. The NAD+-Dependent Family of Sirtuins in Cerebral Ischemia and Preconditioning. Antioxidants Redox Signal. 2018, 28, 691–710. [Google Scholar] [CrossRef]
- Tomizawa, M.; Shinozaki, F.; Fugo, K.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Kishimoto, T.; Ishige, N. Anti-mitochondrial M2 antibody-positive autoimmune hepatitis. Exp. Ther. Med. 2015, 10, 1419–1422. [Google Scholar] [CrossRef]
- Becker, Y.; Loignon, R.-C.; Julien, A.-S.; Marcoux, G.; Allaeys, I.; Lévesque, T.; Rollet-Labelle, E.; Benk-Fortin, H.; Cloutier, N.; Melki, I.; et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Stefanidis, I. Cytochrome c as a Potentially Clinical Useful Marker of Mitochondrial and Cellular Damage. Front. Immunol. 2016, 7, 279. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; Vaishnav, A.; Edwards, B.F.; Grossman, L.I.; Sanderson, T.H.; Lee, I.; Hüttemann, M. Tissue-Specific Regulation of Cytochrome c by Post-Translational Modifications: Respiration, the Mitochondrial Membrane Potential, ROS, and Apoptosis. FASEB J. 2019, 33(2):1540-1553.
- González-Arzola, K.; Velázquez-Cruz, A.; Guerra-Castellano, A.; Casado-Combreras, M. .; Pérez-Mejías, G.; Díaz-Quintana, A.; Díaz-Moreno, I.; De la Rosa, M.. New moonlighting functions of mitochondrial cytochrome c in the cytoplasm and nucleus. FEBS Lett. 2019, 593, 3101–3119. [Google Scholar] [CrossRef]
- Oh, M.; Kim, S.A.; Yoo, H.J. Higher Lactate Level and Lactate-to-Pyruvate Ratio in Autism Spectrum Disorder. Exp. Neurobiol. 2020, 29, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Shayota, B.J. Biomarkers of mitochondrial disorders. Neurotherapeutics 2024, 21, e00325. [Google Scholar] [CrossRef] [PubMed]
- Hubens, W.; Vallbona-Garcia, A.; de Coo, I.; van Tienen, F.; Webers, C.; Smeets, H.; Gorgels, T. Blood biomarkers for assessment of mitochondrial dysfunction: An expert review. Mitochondrion 2022, 62, 187–204. [Google Scholar] [CrossRef]
- McClintock, C.R.; Mulholland, N.; Krasnodembskaya, A.D. Biomarkers of mitochondrial dysfunction in acute respiratory distress syndrome: A systematic review and meta-analysis. Front. Med. 2022, 9, 1011819. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]
- Sharma, S.; Black, S.M. Carnitine Homeostasis, Mitochondrial Function, and Cardiovascular Disease. Drug Discov Today Dis Mech. 2009, 6(1-4):e31-e39.
- McCann, M.R.; De la Rosa, M.V.G.; Rosania, G.R.; Stringer, K.A. L-Carnitine and Acylcarnitines: Mitochondrial Biomarkers for Precision Medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef]
- Liew, G.; Tse, B.; Ho, I.-V.; Joachim, N.; White, A.; Pickford, R.; Maltby, D.; Gopinath, B.; Mitchell, P.; Crossett, B. Acylcarnitine Abnormalities Implicate Mitochondrial Dysfunction in Patients With Neovascular Age-Related Macular Degeneration. Investig. Opthalmology Vis. Sci. 2020, 61, 32–32. [Google Scholar] [CrossRef]
- Nickel, K.; Menke, M.; Endres, D.; Runge, K.; Tucci, S.; Schumann, A.; Domschke, K.; van Elst, L.T.; Maier, S. Altered markers of mitochondrial function in adults with autism spectrum disorder. Autism Res. 2023, 16, 2125–2138. [Google Scholar] [CrossRef]
- Hamilton, R.T.; Walsh, M.E.; Van Remmen, H. Mouse Models of Oxidative Stress Indicate a Role for Modulating Healthy Aging. J Clin Exp Pathol. 2012, 4:005.
- Hibino, M.; Maeki, M.; Tokeshi, M.; Ishitsuka, Y.; Harashima, H.; Yamada, Y. A system that delivers an antioxidant to mitochondria for the treatment of drug-induced liver injury. Sci. Rep. 2023, 13, 1–13. [Google Scholar] [CrossRef]
- Chen, T.-H.; Wang, H.-C.; Chang, C.-J.; Lee, S.-Y. Mitochondrial Glutathione in Cellular Redox Homeostasis and Disease Manifestation. Int. J. Mol. Sci. 2024, 25, 1314. [Google Scholar] [CrossRef]
- Ribas, V.; García -Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial Electron Transport Chain, ROS Generation and Uncoupling (Review). Int J Mol Med. 2019, 44(1):3-15.
- Hass, D.T.; Barnstable, C.J. Uncoupling proteins in the mitochondrial defense against oxidative stress. Prog. Retin. Eye Res. 2021, 83, 100941–100941. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Odnokoz, O.V.; Pavlov, S.V.; Belenicheva, O.I.; Polyakova, E.N. The neuroprotective activity of tamoxifen and tibolone during glutathione depletion in vitro. Neurochem. J. 2012, 6, 202–212. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordero, S.; Kirwan, R.; Noguera-Julian, A.; Cardellach, F.; Fortuny, C.; Morén, C. A Mitocentric View of the Main Bacterial and Parasitic Infectious Diseases in the Pediatric Population. Int. J. Mol. Sci. 2021, 22, 3272. [Google Scholar] [CrossRef]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–6. [Google Scholar] [CrossRef]
- Ghasemi, M.; Mayasi, Y.; Hannoun, A.; Eslami, S.M.; Carandang, R. Nitric Oxide and Mitochondrial Function in Neurological Diseases. Neuroscience 2018, 376, 48–71. [Google Scholar] [CrossRef]
- Belenichev, I.; Gorbachova, S.; Pavlov, S.; Bukhtiyarova, N.; Puzyrenko, A.; Brek, O. NEUROCHEMICAL STATUS OF NITRIC OXIDE IN THE SETTINGS OF THE NORM, ISHEMIC EVENT OF CENTRAL NERVOUS SYSTEM, AND PHARMACOLOGICAL BN INTERVENTION. . 2021, 169–176. [Google Scholar]
- Mokhtari, B.; Yavari, R.; Badalzadeh, R.; Mahmoodpoor, A. An Overview on Mitochondrial-Based Therapies in Sepsis-Related Myocardial Dysfunction: Mitochondrial Transplantation as a Promising Approach. Can. J. Infect. Dis. Med Microbiol. 2022, 2022, 1–17. [Google Scholar] [CrossRef]
- Martemucci, G.; Portincasa, P.; Di Ciaula, A.; Mariano, M.; Centonze, V.; D’alessandro, A.G. Oxidative stress, aging, antioxidant supplementation and their impact on human health: An overview. Mech. Ageing Dev. 2022, 206, 111707. [Google Scholar] [CrossRef]
- Yang, J.; Luo, J.; Tian, X.; Zhao, Y.; Li, Y.; Wu, X. Progress in Understanding Oxidative Stress, Aging, and Aging-Related Diseases. Antioxidants 2024, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P. Kinetics and Mechanisms of Thiol–Disulfide Exchange Covering Direct Substitution and Thiol Oxidation-Mediated Pathways. Antioxidants Redox Signal. 2013, 18, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.L.; Subramanian, P.; Bu, G.; Di Polo, A.; Golde, T.E.; Bovenkamp, D.E. Common Features of Neurodegenerative Disease: Exploring the Brain-Eye Connection and Beyond (Part 1): The 2021 Pre-Symposium of the 15th International Conference on Alzheimer's and Parkinson's Diseases. Mol Neurodegener. 2022, 17(1):68.
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Dietz, J.V.; Fox, J.L.; Khalimonchuk, O. Down the Iron Path: Mitochondrial Iron Homeostasis and Beyond. Cells 2021, 10, 2198. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Norambuena, J.; Flores, R.; Cárdenas, J.P.; Quatrini, R.; Chávez, R.; Levicán, G. Thiol/Disulfide System Plays a Crucial Role in Redox Protection in the Acidophilic Iron-Oxidizing Bacterium Leptospirillum ferriphilum. PLOS ONE 2012, 7, e44576. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Gorbacheva, S.V.; Demchenko, A.V.; Bukhtiyarova, N.V. The thiol-disulfide balance and the nitric oxide system in the brain tissue of rats subjected to experimental acute impairment of cerebral blood flow: The therapeutic effects of nootropic drugs. Neurochem. J. 2014, 8, 24–27. [Google Scholar] [CrossRef]
- Roede, J.R.; Jones, D.P. Reactive species and mitochondrial dysfunction: Mechanistic significance of 4-hydroxynonenal. Environ. Mol. Mutagen. 2010, 51, 380–390. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: A molecular target for amyotrophic lateral sclerosis therapy. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2009, 1802, 186–197. [Google Scholar] [CrossRef]
- Endlicher, R.; Drahota, Z.; Štefková, K.; Červinková, Z.; Kučera, O. The Mitochondrial Permeability Transition Pore-Current Knowledge of Its Structure, Function, and Regulation, and Optimized Methods for Evaluating Its Functional State. Cells. 2023, 12(9):1273.
- Zong, L.; Liang, Z. Apoptosis-inducing factor: a mitochondrial protein associated with metabolic diseases—a narrative review. Cardiovasc. Diagn. Ther. 2023, 12, 609–622. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The Role of Mitochondria in Apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Ademowo, O.S.; Dias, H.K.I.; Burton, D.G.A.; Griffiths, H.R. Lipid (Per)Oxidation in Mitochondria: An Emerging Target in the Ageing Process? Biogerontology. 2017, 18(6):859-879.
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; A Beck, M.; A Brown, D.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. Int. Rev. J. 2018, 9, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Fàbrega, L.; Fernández-Checa, J.C.; de la Rosa, L.C.; Garcia-Ruiz, C. Impact of mitochondrial lipid alterations on liver disease mechanisms and progression. 2024, 3, 382–413. [CrossRef]
- Zhao, J.; Li, J.; Li, G.; Chen, M. The role of mitochondria-associated membranes mediated ROS on NLRP3 inflammasome in cardiovascular diseases. Front. Cardiovasc. Med. 2022, 9, 1059576. [Google Scholar] [CrossRef]
- Lagouge, M.; Larsson, N. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.; Medeiros, M.H.; Di Mascio, P.; Wagner, J.R. Formation and repair of oxidatively generated damage in cellular DNA. Free. Radic. Biol. Med. 2017, 107, 13–34. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.-M. 8-oxoguanine and 8-oxodeoxyguanosine Biomarkers of Oxidative DNA Damage: A Review on HPLC–ECD Determination. Molecules 2022, 27, 1620. [Google Scholar] [CrossRef]
- Weimann, A.; Belling, D.; Poulsen, H.E. Quantification of 8-oxo-guanine and guanine as the nucleobase, nucleoside and deoxynucleoside forms in human urine by high-performance liquid chromatography-electrospray tandem mass spectrometry. Nucleic Acids Res. 2002, 30, 7e–7. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2017, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. The role of poly(ADP-ribosyl)ation in DNA damage response and cancer chemotherapy. Oncogene 2014, 34, 3349–3356. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Dawson, V. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia–reperfusion. Trends Pharmacol. Sci. 1998, 19, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Burgess, J.T.; O’byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S. Mitochondrial signal transduction. Cell Metab. 2022, 34, 1620–1653. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Matuz-Mares, D.; González-Andrade, M.; Araiza-Villanueva, M.G.; Vilchis-Landeros, M.M.; Vázquez-Meza, H. Mitochondrial Calcium: Effects of Its Imbalance in Disease. Antioxidants 2022, 11, 801. [Google Scholar] [CrossRef]
- Moon, D.-O. Calcium’s Role in Orchestrating Cancer Apoptosis: Mitochondrial-Centric Perspective. Int. J. Mol. Sci. 2023, 24, 8982. [Google Scholar] [CrossRef]
- Callens, M.; Kraskovskaya, N.; Derevtsova, K.; Annaert, W.; Bultynck, G.; Bezprozvanny, I.; Vervliet, T. The Role of Bcl-2 Proteins in Modulating Neuronal Ca2+ Signaling in Health and in Alzheimer's Disease. Biochim Biophys Acta Mol Cell Res. 2021, 1868(6):118997.
- Duchen, M.R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int J Mol Sci. 2021, 22(18):9821.
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dalangin, R.; Kim, A.; Campbell, R.E. The Role of Amino Acids in Neurotransmission and Fluorescent Tools for Their Detection. Int. J. Mol. Sci. 2020, 21, 6197. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; Forcaia, G.; Riboni, L.; Ranzato, E.; Sancini, G.; Ambrosone, L.; D'Angelo, E.; Guerra, G.; Moccia, F. Glutamate Triggers Intracellular Ca2+ Oscillations and Nitric Oxide Release by Inducing NAADP- and InsP3-Dependent Ca2+ Release in Mouse Brain Endothelial Cells. J Cell Physiol. 2019, 234(4):3538-3554.
- Lee, H.K.; Choi, S.S.; Han, E.J.; Han, K.J.; Suh, H.W. Role of Glutamate Receptors and an On-Going Protein Synthesis in the Regulation of Phosphorylation of Ca2+/Calmodulin-Dependent Protein Kinase II in the CA3 Hippocampal Region in Mice Administered With Kainic Acid Intracerebroventricularly. Neurosci Lett. 2003, 348(2):93-96.
- Zhao, Y.; Xiong, W.; Li, C.; Zhao, R.; Lu, H.; Song, S.; Zhou, Y.; Hu, Y.; Shi, B.; Ge, J. Hypoxia-induced signaling in the cardiovascular system: pathogenesis and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–42. [Google Scholar] [CrossRef]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Futur. Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef]
- Nicolson, G.L.; de Mattos, G.F.; Ash, M.; Settineri, R.; Escribá, P.V. Fundamentals of Membrane Lipid Replacement: A Natural Medicine Approach to Repairing Cellular Membranes and Reducing Fatigue, Pain, and Other Symptoms While Restoring Function in Chronic Illnesses and Aging. Membranes 2021, 11, 944. [Google Scholar] [CrossRef]
- Szymański, J.; Janikiewicz, J.; Michalska, B.; Patalas-Krawczyk, P.; Perrone, M.; Ziółkowski, W.; Duszyński, J.; Pinton, P.; Dobrzyń, A.; Więckowski, M.R. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int. J. Mol. Sci. 2017, 18, 1576. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F.; Bartoszewski, R. The Role of the Hypoxia-Related Unfolded Protein Response (UPR) in the Tumor Microenvironment. Cancers 2022, 14, 4870. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–40. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Zhang, X.; Laubry, L.; Brunetti, J.; Koenig, S.; Wang, X.; Castelbou, C.; Hetz, C.; Liu, Y.; Frieden, M.; et al. The ER stress sensor IRE1 interacts with STIM1 to promote store-operated calcium entry, T cell activation, and muscular differentiation. Cell Rep. 2023, 42, 113540. [Google Scholar] [CrossRef]
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 18. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Münch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2017, 19, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, I.G.; Park, C.V.; I Yemm, A.; Kenneth, N.S. PERK/eIF2α signaling inhibits HIF-induced gene expression during the unfolded protein response via YB1-dependent regulation of HIF1α translation. Nucleic Acids Res. 2018, 46, 3878–3890. [Google Scholar] [CrossRef] [PubMed]
- Rojas, E.Y.L.G.; Villanueva, C.; Bond, R.A. Hypoxia Inducible Factors as Central Players in the Pathogenesis and Pathophysiology of Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- Rivers, R.J.; Meininger, C.J. The Tissue Response to Hypoxia: How Therapeutic Carbon Dioxide Moves the Response toward Homeostasis and Away from Instability. Int. J. Mol. Sci. 2023, 24, 5181. [Google Scholar] [CrossRef]
- Della Rocca, Y.; Fonticoli, L.; Rajan, T.S.; Trubiani, O.; Caputi, S.; Diomede, F.; Pizzicannella, J.; Marconi, G.D. Hypoxia: molecular pathophysiological mechanisms in human diseases. J. Physiol. Biochem. 2022, 78, 739–752. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, W.; Li, C.; Zhao, R.; Lu, H.; Song, S.; Zhou, Y.; Hu, Y.; Shi, B.; Ge, J. Hypoxia-induced signaling in the cardiovascular system: pathogenesis and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 1–42. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, Y.; Wang, D.; Zhang, X.; Li, Y.; Wang, D.; Liang, Y.; Wang, J.; Zheng, L.; Song, H.; et al. Metabolite and protein shifts in mature erythrocyte under hypoxia. iScience 2024, 27, 109315. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Chandel, N.S.; Simon, M.C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 2020, 21, 268–283. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, F.; Yang, H.; Wang, Z. Action Sites and Clinical Application of HIF-1α Inhibitors. Molecules 2022, 27, 3426. [Google Scholar] [CrossRef] [PubMed]
- Belapurkar, R.; Pfisterer, M.; Dreute, J.; Werner, S.; Zukunft, S.; Fleming, I.; Kracht, M.; Schmitz, M.L. A transient increase of HIF-1α during the G1 phase (G1-HIF) ensures cell survival under nutritional stress. Cell Death Dis. 2023, 14, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Qannita, R.A.; Alalami, A.I.; Harb, A.A.; Aleidi, S.M.; Taneera, J.; Abu-Gharbieh, E.; El-Huneidi, W.; Saleh, M.A.; Alzoubi, K.H.; Semreen, M.H.; et al. Targeting Hypoxia-Inducible Factor-1 (HIF-1) in Cancer: Emerging Therapeutic Strategies and Pathway Regulation. Pharmaceuticals 2024, 17, 195. [Google Scholar] [CrossRef]
- Koyasu, S.; Kobayashi, M.; Goto, Y.; Hiraoka, M.; Harada, H. Regulatory mechanisms of hypoxia-inducible factor 1 activity: Two decades of knowledge. Cancer Sci. 2018, 109, 560–571. [Google Scholar] [CrossRef]
- Gaete, D.; Rodriguez, D.; Watts, D.; Sormendi, S.; Chavakis, T.; Wielockx, B. HIF-Prolyl Hydroxylase Domain Proteins (PHDs) in Cancer-Potential Targets for Anti-Tumor Therapy? Cancers (Basel). 2021, 13(5):988.
- Hirota, K. HIF-α Prolyl Hydroxylase Inhibitors and Their Implications for Biomedicine: A Comprehensive Review. Biomedicines 2021, 9, 468. [Google Scholar] [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and its Potential for Therapeutic Intervention in Malignancy and Ischemia. 2007, 80, 51–60.
- Marsboom, G.; Toth, P.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.-H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-Related Protein 1–Mediated Mitochondrial Mitotic Fission Permits Hyperproliferation of Vascular Smooth Muscle Cells and Offers a Novel Therapeutic Target in Pulmonary Hypertension. Clin. Trans. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef]
- Kirito, K.; Hu, Y.; Komatsu, N. HIF-1 prevents the overproduction of mitochondrial ROS after cytokine stimulation through induction of PDK-Cell Cycle 2009, 8, 2844–2849. [CrossRef]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1alpha-Dependent Suppression of Hypoxia-Induced Apoptosis in Squamous Cell Carcinoma Cells. Cancer Sci. 2005, 96(7):394-402.
- Mialet-Perez, J.; Belaidi, E. Interplay between hypoxia inducible Factor-1 and mitochondria in cardiac diseases. Free. Radic. Biol. Med. 2024, 221, 13–22. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, L.; Peng, R. Hypoxia-Inducible Factor 1 and Mitochondria: An Intimate Connection. Biomolecules 2022, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: the prospect of using HSP70 modulators. Front. Cell. Neurosci. 2023, 17. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.B.; Krueger-Naug, A.M.; Clarke, D.B.; Arrigo, A.-P.; Currie, R.W. The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int. J. Hyperth. 2005, 21, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fernández, M.R.; Gragera, M.; Ochoa-Ibarrola, L.; Quintana-Gallardo, L.; Valpuesta, J.M. Hsp70 – a master regulator in protein degradation. FEBS Lett. 2017, 591, 2648–2660. [Google Scholar] [CrossRef]
- Tsuchida, S.; Arai, Y.; Takahashi, K.A.; Kishida, T.; Terauchi, R.; Honjo, K.; Nakagawa, S.; Inoue, H.; Ikoma, K.; Ueshima, K.; Matsuki, T.; Mazda, O.; Kubo, T. HIF-1α-Induced HSP70 Regulates Anabolic Responses in Articular Chondrocytes Under Hypoxic Conditions. J Orthop Res. 2014, 32(8):975-980.
- Belenichev, I.F.; Kolesnik, Y.M.; Pavlov, S.V.; Sokolik, E.P.; Bukhtiyarova, N.V. Malate-aspartate shunt in neuronal adaptation to ischemic conditions: Molecular-biochemical mechanisms of activation and regulation. Neurochem. J. 2012, 6, 22–28. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Kolesnik, Y.M.; Pavlov, S.V.; Sokolik, E.P.; Bukhtiyarova, N.V. Disturbance of HSP70 chaperone activity is a possible mechanism of mitochondrial dysfunction. Neurochem. J. 2011, 5, 251–256. [Google Scholar] [CrossRef]
- Aliyeva, O.; Belenichev, I.; Popazova, O. Modulation of Hsp70 in the Pharmacological Correction of Nervous System Disorders after Prenatal Hypoxia. International Electronic Conference on Biomedicines. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; p. 39.
- Xu, L.; Voloboueva, L.A.; Ouyang, Y.; Emery, J.F.; Giffard, R.G. Overexpression of Mitochondrial Hsp70/Hsp75 in Rat Brain Protects Mitochondria, Reduces Oxidative Stress, and Protects From Focal Ischemia. J Cereb Blood Flow Metab. 2009, 29(2):365-374.
- Zatsepina, O.G.; Evgen’ev, M.B.; Garbuz, D.G. Role of a Heat Shock Transcription Factor and the Major Heat Shock Protein Hsp70 in Memory Formation and Neuroprotection. Cells 2021, 10, 1638. [Google Scholar] [CrossRef]
- Lyon, M.S.; Milligan, C. Extracellular heat shock proteins in neurodegenerative diseases: New perspectives. Neurosci. Lett. 2019, 711, 134462. [Google Scholar] [CrossRef]
- Dudeja, V.; Mujumdar, N.; Phillips, P.; Chugh, R.; Borja–Cacho, D.; Dawra, R.K.; Vickers, S.M.; Saluja, A.K. Heat Shock Protein 70 Inhibits Apoptosis in Cancer Cells Through Simultaneous and Independent Mechanisms. Gastroenterology 2009, 136, 1772–1782. [Google Scholar] [CrossRef]
- Zhao, K.; Zhou, G.; Liu, Y.; Zhang, J.; Chen, Y.; Liu, L.; Zhang, G. HSP70 Family in Cancer: Signaling Mechanisms and Therapeutic Advances. Biomolecules 2023, 13, 601. [Google Scholar] [CrossRef]
- Nunes, K.P.; de Oliveira, A.A. HSP70: From Signaling Mechanisms to Therapeutics. Biomolecules. 2023, 13, 1141. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, Y.; Gorshkov, B.; Haigh, S.; Bordan, Z.; Weintraub, D.; Rudic, R.D.; Chakraborty, T.; Barman, S.A.; Verin, A.D.; et al. Hsp70 Suppresses Mitochondrial Reactive Oxygen Species and Preserves Pulmonary Microvascular Barrier Integrity Following Exposure to Bacterial Toxins. Front. Immunol. 2018, 9, 1309. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Bath, S.; Marsden, V.S.; Huang, D.C.S.; Metcalf, D.; Harris, A.W.; Strasser, A. FADD and caspase-8 are required for cytokine-induced proliferation of hemopoietic progenitor cells. Blood 2005, 106, 1581–1589. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Lien, C.-I.; Tseng, Y.-C.; Tu, Y.-F.; Kulczyk, A.W.; Lu, Y.-C.; Wang, Y.-T.; Su, T.-W.; Hsu, L.-C.; Lo, Y.-C.; et al. Deciphering DED assembly mechanisms in FADD-procaspase-8-cFLIP complexes regulating apoptosis. Nat. Commun. 2024, 15, 1–18. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Chen, J.D. Daxx Silencing Sensitizes Cells to Multiple Apoptotic Pathways. Mol. Cell. Biol. 2003, 23, 7108–7121. [Google Scholar] [CrossRef]
- Seyrek, K.; Espe, J.; Reiss, E.; Lavrik, I.N. The Crosstalk of Apoptotic and Non-Apoptotic Signaling in CD95 System. Cells 2024, 13, 1814. [Google Scholar] [CrossRef]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Zhang, K.; Zhai, R.; Xue, T.; Xu, X.; Ren, Y.; Ma, M.; Shi, F.; Wang, H.; Wang, N.; Zhou, F. HSP70 regulates cell proliferation and apoptosis in actinomycin-D-treated lung cancer cells. Transl. Cancer Res. 2020, 9, 1167–1173. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome c Promotes Caspase-9 Activation by Inducing Nucleotide Binding to Apaf-J. Biol. Chem. 2000, 275, 31199–31203. [Google Scholar] [CrossRef]
- Merlin, J.P.J.; Crous, A.; Abrahamse, H. Combining Photodynamic Therapy and Targeted Drug Delivery Systems: Enhancing Mitochondrial Toxicity for Improved Cancer Outcomes. Int. J. Mol. Sci. 2024, 25, 10796. [Google Scholar] [CrossRef]
- Li, C.-Y.; Lee, J.-S.; Ko, Y.-G.; Kim, J.-I.; Seo, J.-S. Heat Shock Protein 70 Inhibits Apoptosis Downstream of Cytochrome c Release and Upstream of Caspase-3 Activation. J. Biol. Chem. 2000, 275, 25665–25671. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Bratton, S.B. Caspase-9 swings both ways in the apoptosome. Mol. Cell. Oncol. 2016, 4, e1281865. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Wang, L.; Du, F. Novel Small Molecules Relieve Prothymosin Alpha-Mediated Inhibition of Apoptosome Formation by Blocking Its Interaction With Apaf-Biochemistry. 2010, 49(9):1923-1930.
- Leu, J.I.-J.; Barnoud, T.; Zhang, G.; Tian, T.; Wei, Z.; Herlyn, M.; Murphy, M.E.; George, D.L. Inhibition of stress-inducible HSP70 impairs mitochondrial proteostasis and function. Oncotarget 2017, 8, 45656–45669. [Google Scholar] [CrossRef] [PubMed]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-Encyclopedia 2022, 2, 1624–1636. [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxidative Med. Cell. Longev. 2019, 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Endlicher, R.; Drahota, Z.; Štefková, K.; Červinková, Z.; Kučera, O. The Mitochondrial Permeability Transition Pore-Current Knowledge of Its Structure, Function, and Regulation, and Optimized Methods for Evaluating Its Functional State. Cells. 2023, 12(9):1273.
- Kinnally, K.W.; Zorov, D.B.; Antonenko, Y.N.; Snyder, S.H.; McEnery, M.W.; Tedeschi, H. Mitochondrial benzodiazepine receptor linked to inner membrane ion channels by nanomolar actions of ligands. Proc. Natl. Acad. Sci. 1993, 90, 1374–1378. [Google Scholar] [CrossRef]
- Servida, F.; Lecis, D.; Scavullo, C.; Drago, C.; Seneci, P.; Carlo-Stella, C.; Manzoni, L.; Polli, E.; Deliliers, G.L.; Delia, D.; et al. Novel second mitochondria-derived activator of caspases (Smac) mimetic compounds sensitize human leukemic cell lines to conventional chemotherapeutic drug-induced and death receptor-mediated apoptosis. Investig. New Drugs 2010, 29, 1264–1275. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704–105704. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Goldstein, J.C.; von Ahsen, O.; Schuler, M.; Newmeyer, D.D.; Green, D.R. Cytochrome C Maintains Mitochondrial Transmembrane Potential and Atp Generation after Outer Mitochondrial Membrane Permeabilization during the Apoptotic Process. J. Cell Biol. 2001, 153, 319–328. [Google Scholar] [CrossRef]
- Barroso, G.; Morshedi, M.; Oehninger, S. Analysis of DNA fragmentation, plasma membrane translocation of phosphatidylserine and oxidative stress in human spermatozoa. Hum. Reprod. 2000, 15, 1338–1344. [Google Scholar] [CrossRef]
- Ramos, M.F.; Baker, J.; Atzpodien, E.-A.; Bach, U.; Brassard, J.; Cartwright, J.; Farman, C.; Fishman, C.; Jacobsen, M.; Junker-Walker, U.; et al. Nonproliferative and Proliferative Lesions of the Rat and Mouse Special Sense Organs (Ocular eye and glands], Olfactory and Otic). J. Toxicol. Pathol. 2018, 31, 97S–214S. [Google Scholar] [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.; Tamrakar, P.; Rosenthal, R.E.; Fiskum, G. Calcium Uptake and Cytochrome c Release From Normal and Ischemic Brain Mitochondria. Neurochem Int. 2018, 117:15-22.
- Hardwick, J.M.; Soane, L. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722–a008722. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Hayashi, T.; Su, T.-P.; Betenbaugh, M.J. Bcl-2 family in inter-organelle modulation of calcium signaling; roles in bioenergetics and cell survival. J. Bioenerg. Biomembr. 2013, 46, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Saddam; Paul, S.K.; Habib, M.A.; Fahim, A.; Mimi, A.; Islam, S.; Paul, B.; Helal, M.U. Emerging biomarkers and potential therapeutics of the BCL-2 protein family: the apoptotic and anti-apoptotic context. Egypt. J. Med Hum. Genet. 2024, 25, 1–28. [Google Scholar] [CrossRef]
- Green, D.R. The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family. Cold Spring Harb. Perspect. Biol. 2022, 14, a041046. [Google Scholar] [CrossRef]
- Nguyen, D.; Osterlund, E.; Kale, J.; Andrews, D.W. The C-terminal sequences of Bcl-2 family proteins mediate interactions that regulate cell death. Biochem. J. 2024, 481, 903–922. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Bahatyrevich-Kharitonik, B.; Medina-Guzman, R.; Flores-Cortes, A.; García-Cruzado, M.; Kavanagh, E.; Burguillos, M.A. Cell Death Related Proteins Beyond Apoptosis in the CNS. Front. Cell Dev. Biol. 2022, 9, 825747. [Google Scholar] [CrossRef] [PubMed]
- Wolf, P.; Schoeniger, A.; Edlich, F. Pro-apoptotic complexes of BAX and BAK on the outer mitochondrial membrane. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2022, 1869, 119317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Yue, G.; Zhao, Y. Energy Metabolism Disturbance in Migraine: From a Mitochondrial Point of View. Front. Physiol. 2023, 14:1133528.
- ileikytė, J.; Forte, M. The Mitochondrial Permeability Transition in Mitochondrial Disorders. Oxid Med Cell Longev. 2019, 2019:3403075.
- Trindade, D.; Pereira, C.; Chaves, S.R.; Manon, S.; Corte-Real, M.; Sousa, M.J. VDAC regulates AAC-mediated apoptosis and cytochrome c release in yeast. Microb. Cell 2016, 3, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Gerle, C.; Halestrap, A.P.; Jonas, E.A.; Karch, J.; Mnatsakanyan, N.; Pavlov, E.; Sheu, S.-S.; Soukas, A.A. Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023, 30, 1869–1885. [Google Scholar] [CrossRef]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2006, 1757, 639–647. [Google Scholar] [CrossRef]
- Naumova, N.; Šachl, R. Regulation of Cell Death by Mitochondrial Transport Systems of Calcium and Bcl-2 Proteins. Membranes 2020, 10, 299. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Maes, M.E.; Schlamp, C.L.; Nickells, R.W. BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog. Retin. Eye Res. 2017, 57, 1–25. [Google Scholar] [CrossRef]
- Kotrasová, V.; Keresztesová, B.; Ondrovičová, G.; Bauer, J.A.; Havalová, H.; Pevala, V.; Kutejová, E.; Kunová, N. Mitochondrial Kinases and the Role of Mitochondrial Protein Phosphorylation in Health and Disease. Life 2021, 11, 82. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Pahlavani, H.A. Exercise-induced signaling pathways to counteracting cardiac apoptotic processes. Front. Cell Dev. Biol. 2022, 10, 950927. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How Does p53 Induce Apoptosis and How Does This Relate to p53-Mediated Tumour Suppression? Cell Death Differ. 2018, 25(1):104-113.
- la Cour, J.M.; Høj, B.R.; Mollerup, J.; Simon, R.; Sauter, G.; Berchtold, M.W. The apoptosis linked gene ALG-2 is dysregulated in tumors of various origin and contributes to cancer cell viability. Mol. Oncol. 2007, 1, 431–439. [Google Scholar] [CrossRef]
- McCullough, J.; Fisher, R.D.; Whitby, F.G.; Sundquist, W.I.; Hill, C.P. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc. Natl. Acad. Sci. 2008, 105, 7687–7691. [Google Scholar] [CrossRef]
- Zanatta, D.; Betanzos, A.; Azuara-Liceaga, E.; Montaño, S.; Orozco, E. Entamoeba histolytica: EhADH, an Alix Protein, Participates in Several Virulence Events through Its Different Domains. Int. J. Mol. Sci. 2024, 25, 7609. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706–a008706. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Li, Y.; Rasheed, M.; Liu, J.; Chen, Z.; Deng, Y. Deciphering the Molecular Nexus: An In-Depth Review of Mitochondrial Pathways and Their Role in Cell Death Crosstalk. Cells 2024, 13, 863. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Dadsena, S.; García-Sáez, A.J. Mitochondrial pores at the crossroad between cell death and inflammatory signaling. 2023, 83, 843–856. [CrossRef]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Spagnoletta, A.; Oppedisano, F. Inflammation, Mitochondria and Natural Compounds Together in the Circle of Trust. Int. J. Mol. Sci. 2023, 24, 6106. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.; Garcia-Dorado, D.; Action, E.-C.C. Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J. Cell. Mol. Med. 2020, 24, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, D.J.; Harata, M.; Murphy, E.; Karch, J. Mitochondrial permeability transition pore-dependent necrosis. 2022, 174, 47–55. [CrossRef]
- Javadov, S.; Chapa-Dubocq, X.; Makarov, V. Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 2018, 38, 58–70. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Wei, S.; Jo, Y.; Zhang, Y.; Park, W.; Gariani, K.; Oh, C.-M.; Kim, H.H.; Ha, K.-T.; Park, K.S.; et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp. Mol. Med. 2023, 55, 1595–1619. [Google Scholar] [CrossRef]
- Miller, M.A.; Zachary, J.F. Mechanisms and Morphology of Cellular Injury, Adaptation, and Death 11 For a glossary of abbreviations and terms used in this chapter see E-Glossary 1-1. Pathol. Basis Vet. Dis. 2017, 2–43.e19. [CrossRef]
- Khan, T.; Waseem, R.; Zehra, Z.; Aiman, A.; Bhardwaj, P.; Ansari, J.; Hassan, I.; Islam, A. Mitochondrial Dysfunction: Pathophysiology and Mitochondria-Targeted Drug Delivery Approaches. Pharmaceutics 2022, 14, 2657. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, H.; Sandai, D.; Zhao, R.; Bai, J.; Wang, Y.; Wang, Y.; Zhang, Z.; Zhang, H.-L.; Song, Z.-J. ATP-induced cell death: a novel hypothesis for osteoporosis. Front. Cell Dev. Biol. 2023, 11, 1324213. [Google Scholar] [CrossRef]
- Imamura, H.; Sakamoto, S.; Yoshida, T.; Matsui, Y.; Penuela, S.; Laird, D.W.; Mizukami, S.; Kikuchi, K.; Kakizuka, A.; Biology, C.; et al. Single-cell dynamics of pannexin-1-facilitated programmed ATP loss during apoptosis. eLife 2020, 9. [Google Scholar] [CrossRef]
- Bartman, S.; Coppotelli, G.; Ross, J.M. Mitochondrial Dysfunction: A Key Player in Brain Aging and Diseases. Curr. Issues Mol. Biol. 2024, 46, 1987–2026. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-Demiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. EBioMedicine 2021, 65, 103244. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Han, Y.; He, Y.; Liu, J.; Wang, Y. Natural compounds targeting mitochondrial dysfunction: emerging therapeutics for target organ damage in hypertension. Front. Pharmacol. 2023, 14, 1209890. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.J.; Hannan, M.A.; Rahman, M.A.; Apostolova, N. Editorial: Novel Pharmacological Approaches Targeting Mitochondrial Dysfunction in Diseases. Front. Pharmacol. 2022, 13:1041576.
- Tinker, R.J.; Lim, A.Z.; Stefanetti, R.J.; McFarland, R. Current and Emerging Clinical Treatment in Mitochondrial Disease. Mol. Diagn. Ther. 2021, 25, 181–206. [Google Scholar] [CrossRef] [PubMed]
- Novikov, V.E.; Levchenkova, O.S.; Ivantsova, E.N.; Vorobieva, V.V. Mitochondrial Dysfunctions and Antihypoxants. Reviews on Clinical Pharmacology and Drug Therapy. 2019, 17(4):31–42.
- Korski, K.I.; Kubli, D.A.; Wang, B.J.; Khalafalla, F.G.; Monsanto, M.M.; Firouzi, F.; Echeagaray, O.H.; Kim, T.; Adamson, R.M.; Dembitsky, W.P.; et al. Hypoxia Prevents Mitochondrial Dysfunction and Senescence in Human c-Kit+ Cardiac Progenitor Cells. STEM CELLS 2019, 37, 555–567. [Google Scholar] [CrossRef]
- Cuvillier, O.; Ader, I.; Bouquerel, P.; Brizuela, L.; Gstalder, C.; Malavaud, B. Hypoxia, Therapeutic Resistance, and Sphingosine 1-Phosphate. Adv Cancer Res. 2013, 117:117–141.
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef]
- Bouhamida, E.; Morciano, G.; Perrone, M.; Kahsay, A.E.; Della Sala, M.; Wieckowski, M.R.; Fiorica, F.; Pinton, P.; Giorgi, C.; Patergnani, S. The Interplay of Hypoxia Signaling on Mitochondrial Dysfunction and Inflammation in Cardiovascular Diseases and Cancer: From Molecular Mechanisms to Therapeutic Approaches. Biology 2022, 11, 300. [Google Scholar] [CrossRef]
- Sifat, A.E.; Nozohouri, S.; Archie, S.R.; Chowdhury, E.A.; Abbruscato, T.J. Brain Energy Metabolism in Ischemic Stroke: Effects of Smoking and Diabetes. Int. J. Mol. Sci. 2022, 23, 8512. [Google Scholar] [CrossRef]
- Martinez, C.S.; Zheng, A.; Xiao, Q. Mitochondrial Reactive Oxygen Species Dysregulation in Heart Failure with Preserved Ejection Fraction: A Fraction of the Whole. Antioxidants 2024, 13, 1330. [Google Scholar] [CrossRef]
- Arnalich-Montiel, A.; Burgos-Santamaría, A.; Pazó-Sayós, L.; Quintana-Villamandos, B. Comprehensive Management of Stroke: From Mechanisms to Therapeutic Approaches. Int. J. Mol. Sci. 2024, 25, 5252. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J. Bioenerg. Biomembr. 2015, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Schulz, J.B.; Giunti, P. Co-Enzyme Q10 and Idebenone Use in Friedreich's Ataxia. J Neurochem. 2013, 126(Suppl 1):125–141.
- Suárez-Rivero, J.M.; Pastor-Maldonado, C.J.; Povea-Cabello, S.; Álvarez-Córdoba, M.; Villalón-García, I.; Munuera-Cabeza, M.; Suárez-Carrillo, A.; Talaverón-Rey, M.; Sánchez-Alcázar, J.A. Coenzyme Q10 Analogues: Benefits and Challenges for Therapeutics. Antioxidants 2021, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Bergamasco, B.; Villardita, C.; Coppi, R. Idebenone in the treatment of multi-infarct dementia: a randomised, double-blind, placebo controlled multicentre trial. Arch. Gerontol. Geriatr. 1992, 15, 271–278. [Google Scholar] [CrossRef]
- Marigliano, V.; Abate, G.; Barbagallo-Sangiorgi, G.; Bartorelli, L.; Capurso, A.; Cucinotta, D.; Cuzzupoli, M.; Senin, U.; Tammaro, A.E.; Fioravanti, M. Randomized, double-blind, placebo controlled, multicentre study of idebenone in patients suffering from multi-infarct dementia. Arch. Gerontol. Geriatr. 1992, 15, 239–248. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Luo, N.; Chen, F.; Zhou, L.; Niu, M.; Kang, W.; Liu, J. Efficacy of idebenone in the Treatment of iRBD into Synucleinopathies (EITRS): rationale, design, and methodology of a randomized, double-blind, multi-center clinical study. Front. Neurol. 2022, 13, 981249. [Google Scholar] [CrossRef]
- Shneyvays, V.; Leshem, D.; Shmist, Y.; Zinman, T.; Shainberg, A. Effects of menadione and its derivative on cultured cardiomyocytes with mitochondrial disorders. J. Mol. Cell. Cardiol. 2005, 39, 149–158. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Hou, X.; Jacob, C. 1,4-Naphthoquinones: From Oxidative Damage to Cellular and Inter-Cellular Signaling. Molecules 2014, 19, 14902–14918. [Google Scholar] [CrossRef]
- Schmidt, C.A. Prescription drugs and mitochondrial metabolism. Biosci. Rep. 2022, 42. [Google Scholar] [CrossRef]
- Tang, Q.; Xie, Y.; Liu, Y.; Zheng, L. Synthesis of mitochondria-targeted menadione cation derivatives: Inhibiting mitochondrial thioredoxin reductase (TrxR2) and inducing apoptosis in MGC-803 cells. Bioorganic Med. Chem. Lett. 2022, 60, 128586. [Google Scholar] [CrossRef]
- Yaneva, Z.; Ivanova, D.; Toneva, M.; Tzanova, M.; Marutsova, V.; Grozeva, N. Menadione Contribution to the In Vitro Radical Scavenging Potential of Phytochemicals Naringenin and Lignin. Int. J. Mol. Sci. 2023, 24, 16268. [Google Scholar] [CrossRef] [PubMed]
- A Merchant, H. The synergistic combination of trimetazidine, hypoxen and L-carnitine in endurance sports. Br. J. Pharm. 2032, 7. [Google Scholar] [CrossRef]
- Görgens, C.; Möller, T.; Guddat, S.; Svambayev, E.; Geyer, H.; Thomas, A.; Thevis, M. Urinary metabolites indicative of the administration of hypoxen monitored by liquid chromatography–high resolution/accurate mass tandem mass spectrometry. Drug Test. Anal. 2024. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K. Extracellular Succinate: A Physiological Messenger and a Pathological Trigger. Int. J. Mol. Sci. 2023, 24, 11165. [Google Scholar] [CrossRef]
- Huang, H.; Li, G.; He, Y.; Chen, J.; Yan, J.; Zhang, Q.; Li, L.; Cai, X. Cellular succinate metabolism and signaling in inflammation: implications for therapeutic intervention. Front. Immunol. 2024, 15, 1404441. [Google Scholar] [CrossRef]
- Zhang, W.; Lang, R. Succinate metabolism: a promising therapeutic target for inflammation, ischemia/reperfusion injury and cancer. Front. Cell Dev. Biol. 2023, 11, 1266973. [Google Scholar] [CrossRef]
- Bielenichev, I.F.; Gorchakova, N.A.; Doroshenko, E.Y.; Samura, I.B.; Ryzhenko, V.P.; Bukhtiiarova, N.V. Use of metabolites, metabolithotropic agents and nutritional supplements in sports and sports medicine: a modern view on the problem. Mod. Med Technol. 2023, 76–88. [Google Scholar] [CrossRef]
- Oguro, H.; Iijima, K.; Takahashi, K.; Nagai, A.; Bokura, H.; Yamaguchi, S.; Kobayashi, S. Successful Treatment with Succinate in a Patient with MELAS. Intern. Med. 2004, 43, 427–431. [Google Scholar] [CrossRef]
- Ehinger, J.K.; Piel, S.; Ford, R.; Karlsson, M.; Sjövall, F.; Frostner, E. .; Morota, S.; Taylor, R.W.; Turnbull, D.; Cornell, C.; et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016, 7, 12317. [Google Scholar] [CrossRef]
- Jędrejko, K.; Catlin, O.; Stewart, T.; Muszyńska, B. Mexidol, Cytoflavin, and succinic acid derivatives as antihypoxic, anti-ischemic metabolic modulators, and ergogenic aids in athletes and consideration of their potential as performance enhancing drugs. Drug Test. Anal. 2024. [Google Scholar] [CrossRef]
- Gorchakova, N.; Belenichev, I.; Harnyk, T.; Shumeiko, O.; Klymenko, O. Membranotropic Action of Phytodrugs. Phytotherapy J. 2023, 4:5–10.
- Liu, X.; Zhao, G.; Sun, S.; Fan, C.; Feng, X.; Xiong, P. Biosynthetic Pathway and Metabolic Engineering of Succinic Acid. Front. Bioeng. Biotechnol. 2022, 10, 843887. [Google Scholar] [CrossRef] [PubMed]
- Pedriali, G.; Ramaccini, D.; Bouhamida, E.; Wieckowski, M.R.; Giorgi, C.; Tremoli, E.; Pinton, P. Perspectives on mitochondrial relevance in cardiac ischemia/reperfusion injury. Front. Cell Dev. Biol. 2022, 10, 1082095. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Vercellino, I.; Sazanov, L.A. The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 2021, 23, 141–161. [Google Scholar] [CrossRef]
- Mansilla, N.; Racca, S.; Gras, D.E.; Gonzalez, D.H.; Welchen, E. The Complexity of Mitochondrial Complex IV: An Update of Cytochrome c Oxidase Biogenesis in Plants. Int. J. Mol. Sci. 2018, 19, 662. [Google Scholar] [CrossRef]
- Skulachev, V.P. Cationic antioxidants as a powerful tool against mitochondrial oxidative stress. Biochem. Biophys. Res. Commun. 2013, 441, 275–279. [Google Scholar] [CrossRef]
- Fock, E.M.; Parnova, R.G. Protective Effect of Mitochondria-Targeted Antioxidants against Inflammatory Response to Lipopolysaccharide Challenge: A Review. Pharmaceutics 2021, 13, 144. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Averina, O.A.; Andreev-Andrievskiy, A.A.; Zinovkin, R.A.; Lyamzaev, K.G.; Marey, M.V.; Egorov, M.V.; Frolova, O.J.; et al. Mitochondrion-targeted antioxidant SkQ1 prevents rapid animal death caused by highly diverse shocks. Sci. Rep. 2023, 13, 1–15. [Google Scholar] [CrossRef]
- Ivanov, B.; Khorobrykh, S. Participation of Photosynthetic Electron Transport in Production and Scavenging of Reactive Oxygen Species. Antioxidants Redox Signal. 2003, 5, 43–53. [Google Scholar] [CrossRef]
- Silachev, D.N.; Plotnikov, E.Y.; Zorova, L.D.; Pevzner, I.B.; Sumbatyan, N.V.; Korshunova, G.A.; Gulyaev, M.V.; Pirogov, Y.A.; Skulachev, V.P.; Zorov, D.B. Neuroprotective Effects of Mitochondria-Targeted Plastoquinone and Thymoquinone in a Rat Model of Brain Ischemia/Reperfusion Injury. Molecules 2015, 20, 14487–14503. [Google Scholar] [CrossRef]
- Zhou, Z.; Austin, G.L.; Young, L.E.A.; Johnson, L.A.; Sun, R. Mitochondrial Metabolism in Major Neurological Diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-Demiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef] [PubMed]
- Baba, S.P.; Bhatnagar, A. Role of thiols in oxidative stress. Curr. Opin. Toxicol. 2018, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kükürt, A.; Gelen, V.; Faruk Başer, Ö.; Deveci, H.A.; Karapehlivan, M. Thiols: Role in Oxidative Stress-Related Disorders. IntechOpen. 2021.
- McCarty, M.F.; DiNicolantonio, J.J.; Lerner, A. A Fundamental Role for Oxidants and Intracellular Calcium Signals in Alzheimer’s Pathogenesis—And How a Comprehensive Antioxidant Strategy May Aid Prevention of This Disorder. Int. J. Mol. Sci. 2021, 22, 2140. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Bystrzycka, W.; Cieloch, A.; Glodkowska-Mrowka, E.; Jankowska-Steifer, E.; Heropolitanska-Pliszka, E.; Skrobot, A.; Muchowicz, A.; Ciepiela, O.; Wachowska, M.; et al. Nitric oxide and peroxynitrite trigger and enhance release of neutrophil extracellular traps. Cell. Mol. Life Sci. 2019, 77, 3059–3075. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, G.; Godínez-Méndez, L.A.; Fafutis-Morris, M.; Delgado-Rizo, V. Effect of Antioxidant Supplementation on NET Formation Induced by LPS In Vitro: The Roles of Vitamins E and C, Glutathione, and N-Acetyl Cysteine. Int. J. Mol. Sci. 2023, 24:13162.
- Gupta, D.S.; Parab, S.B.; Kaur, G. Promising effects of emoxypine and its succinate derivative in the management of various diseases-with insights on recent patent applications. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100121. [Google Scholar] [CrossRef]
- Peresypkina, A.; Pazhinsky, A.; Danilenko, L.; Lugovskoy, S.; Pokrovskii, M.; Beskhmelnitsyna, E.; Solovev, N.; Pobeda, A.; Korokin, M.; Levkova, E.; et al. Retinoprotective Effect of 2-Ethyl-3-hydroxy-6-methylpyridine Nicotinate. Biology 2020, 9, 45. [Google Scholar] [CrossRef]
- Chekman, I.; Belenichev, I.; Yakovleva, I.; Gorchakova, N.; Buhtiyarova, N.; Morguntsova, S.; Brazhko, O.; Levich, S. Influence of mexidol on early genomic response and morphofunctional parameters of brain cortex sensorimotor zone neurons after arteria carotis communis occlusion. Oxid. Antioxidants Med Sci. 2015, 4, 33–38. [Google Scholar] [CrossRef]
- Parkhomenko, D.; Belenichev, I.; Kuchkovskyi, O.; Ryzhenko, V. Characteristics of HIF-1α and HSP70 mRNA Expression, Level, and Interleukins in Experimental Chronic Generalized Periodontitis. Microrna. 2024, 13(2):132–139.
- Alekseeva, T.G.; Loseva, E.V.; Mering, T.A. The effects of Mexidol on the acquisition of food-related conditioned reflexes and synaptic ultrastructure in field Ca1 of the rat hippocampus after single acoustic stimuli with ultrasonic components. Neurosci. Behav. Physiol. 2005, 35, 363–369. [Google Scholar] [CrossRef]
- Belenichev, I.; Ryzhenko, V.; Popazova, O.; Bukhtiyarova, N.; Gorchakova, N.; Oksenych, V.; Kamyshnyi, O. Optimization of the Search for Neuroprotectors among Bioflavonoids. Pharmaceuticals 2024, 17, 877. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Shevchenko, A.O.; Kyryliuk, A.D.; Siusiuka, V.G. Premature Childbirth: Fundamental and Clinical Aspects. LAP LAMBERT Academic Publishing. 2022, 308 p.
- Panov, A.V.; Mayorov, V.I.; Dikalova, A.E.; Dikalov, S.I. Long-Chain and Medium-Chain Fatty Acids in Energy Metabolism of Murine Kidney Mitochondria. Int. J. Mol. Sci. 2022, 24, 379. [Google Scholar] [CrossRef] [PubMed]
- Popazova, O.; Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Oksenych, V.; Kamyshnyi, A. Cardioprotective Activity of Pharmacological Agents Affecting NO Production and Bioavailability in the Early Postnatal Period after Intrauterine Hypoxia in Rats. Biomedicines 2023, 11, 2854. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; He, H.; Yang, W.; Wang, D.; Sui, X.; Sun, Y.; Wang, S.; Yang, Y.; Xiao, Z.; Yang, J.; et al. Novel energy optimizer, meldonium, rapidly restores acute hypobaric hypoxia-induced brain injury by targeting phosphoglycerate kinase Cell Commun. Signal. 2024, 22, 1–17. [Google Scholar] [CrossRef]
- Yang, W.; Lei, X.; Liu, F.; Sui, X.; Yang, Y.; Xiao, Z.; Cui, Z.; Sun, Y.; Yang, J.; Yang, X.; Lin, X.; Bao, Z.; Li, W.; Ma, Y.; Wang, Y.; Luo, Y. Meldonium, as a Potential Neuroprotective Agent, Promotes Neuronal Survival by Protecting Mitochondria in Cerebral Ischemia-Reperfusion Injury. J. Transl. Med. 2024, 22(1):771.
- Bielenichev, I.F.; Vіzіr, V.A.; Mamchur, V.Y.; Kuriata, O.V. Place of tiotriazoline in the gallery of modern metabolitotropic medicines. Zaporozhye Med J. 2019, 118–128. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Feroz, S.; Chekman, I.S.; Nagornaya, E.A.; Gorbacheva, S.V.; Gorchakova, N.A.; et al. Thiol-Disulfide System: Role in Endogenous Cyto- and Organoprotection, Pathways of Pharmacological Modulation. Kyiv: Yuston Publishing House. 2020, 232 p.
- Belenichev, I.F.; Aliyeva, E.G.; Popazova, O.O. Experimental Substantiation of New Target Links in Complex Therapy of Prenatal CNS Damage: Pharmacological Modulation of HSP70-Dependent Mechanisms of Endogenous Neuroprotection. Neurotherapeutics. 2022, 19:1414–1431.
- Popazova, O.; Belenichev, I.; Yadlovskyi, O.; Oksenych, V.; Kamyshnyi, A. Altered Blood Molecular Markers of Cardiovascular Function in Rats after Intrauterine Hypoxia and Drug Therapy. Curr. Issues Mol. Biol. 2023, 45, 8704–8715. [Google Scholar] [CrossRef]
- Belenichev, I.; Kucherenko, L.; Pavlov, S.; Bukhtiyarova, N.; Popazova, O.; Derevianko, N.; Nimenko, G. Therapy of post-COVID-19 syndrome: improving the efficiency and safety of basic metabolic drug treatment with tiazotic acid (thiotriazoline). Pharmacia 2022, 69, 509–516. [Google Scholar] [CrossRef]
- Kamenshchyk, A.; Belenichev, I.; Oksenych, V.; Kamyshnyi, O. Combined Pharmacological Modulation of Translational and Transcriptional Activity Signaling Pathways as a Promising Therapeutic Approach in Children with Myocardial Changes. Biomolecules 2024, 14, 477. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Mazur, I.A.; Abramov, A.V.; Kucherenko, L.I.; Bukhtiyarova, N.V.; Egorov, A.A.; Belenicheva, O.I.; Polyakova, E.N. The endothelium-protective effect of 3-methyl-1,2,4-triazolyl-5-thioacetate (S)-2,6-diaminohexanic acid (lysinium): Effects on the expression of vascular endothelial growth factor (VEGF) and the characteristics of the endotheliocytes of the cerebral vessels of animals with cerebral ischemia. Neurochem. J. 2013, 7, 296–302. [Google Scholar] [CrossRef]
- Belenichev, I.; Kucherenko, L.; Nagornaya, E.; Mazur, I.; Bidnenko, A.; Bukhtiyarova, N.; Avramenko, N. Functional nitric oxide conjugate systems state/restored heart thiols of rats in modeling isadrine-pituitrin's myocardial infarction using metabolite-tropic cardioprotector “Angiolin”. Int. J. Basic Clin. Pharmacol. 2015, 4, 15. [Google Scholar] [CrossRef]
- Aliyeva, O.; Belenichev, I.; Bukhtiyarova, N.; Semenov, D.; Voloshchuk, S. Comparative Assessment of the Effectiveness of HSP70 / HIF-1α System Modulators After Prenatal Hypoxia. Biomed Pharmacol J. 2024, 17(1).
- Belenichev, I.; Aliyeva, O.; Gunina, L.; Bukhtiyarova, N. EVALUATION OF THE EFFICIENCY OF THE NEUROPROTECTIVE DRUGS AFTER PRENATAL HYPOXIA. Fiziolohichnyĭ zhurnal 2023, 69, 43–53. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Aliyeva, E.G.; Kamyshny, O.M.; Bukhtiyarova, N.V.; Ryzhenko, V.P.; Gorchakova, N.O. Pharmacological Modulation of Endogenous Neuroprotection after Experimental Prenatal Hypoxia. Neurochem. J. 2022, 16, 68–75. [Google Scholar] [CrossRef]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem. Int. 2002, 40, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovasc. Res. 2009, 83, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Šileikytė, J.; Blachly-Dyson, E.; Sewell, R.; Carpi, A.; Menabò, R.; Di Lisa, F.; Ricchelli, F.; Bernardi, P.; Forte, M. Regulation of the Mitochondrial Permeability Transition Pore by the Outer Membrane Does Not Involve the Peripheral Benzodiazepine Receptor (Translocator Protein of 18 kDa (TSPO)). J. Biol. Chem. 2014, 289, 13769–13781. [Google Scholar] [CrossRef]
- Sarnowska, A.; Beręsewicz, M.; Zabłocka, B.; Domańska-Janik, K. Diazepam neuroprotection in excitotoxic and oxidative stress involves a mitochondrial mechanism additional to the GABAAR and hypothermic effects. Neurochem. Int. 2009, 55, 164–173. [Google Scholar] [CrossRef]
- Sato, K.; Takayama, K.-I.; Inoue, S. Expression and function of estrogen receptors and estrogen-related receptors in the brain and their association with Alzheimer’s disease. Front. Endocrinol. 2023, 14, 1220150. [Google Scholar] [CrossRef]
- Russell, J.K.; Jones, C.K.; Newhouse, P.A. The Role of Estrogen in Brain and Cognitive Aging. Neurotherapeutics 2019, 16, 649–665. [Google Scholar] [CrossRef]
- Devillers, M.M.; Mhaouty-Kodja, S.; Guigon, C.J. Deciphering the Roles & Regulation of Estradiol Signaling during Female Mini-Puberty: Insights from Mouse Models. Int. J. Mol. Sci. 2022, 23, 13695. [Google Scholar] [CrossRef]
- Del Río, J.P.; Molina, S.; Hidalgo-Lanussa, O.; Garcia-Segura, L.M.; Barreto, G.E. Tibolone as Hormonal Therapy and Neuroprotective Agent. Trends Endocrinol. Metab. 2020, 31, 742–759. [Google Scholar] [CrossRef]
- Novick, A.M.; Scott, A.T.; Epperson, C.N.; Schneck, C.D. Neuropsychiatric effects of tamoxifen: Challenges and opportunities. Front. Neuroendocr. 2020, 59, 100869–100869. [Google Scholar] [CrossRef]
- Lee, E.Y.; Sidoryk, M.; Jiang, H.; Yin, Z.; Aschner, M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J. Neurochem. 2009, 110, 530–544. [Google Scholar] [CrossRef]
- Pavlov, S.V.; Belenichev, I.F. Molecular and biochemical aspects of the neuroprotective effect of the selective estrogen receptor modulator tamoxifen in a model of acute cerebral ischemia. Neurochem. J. 2014, 8, 28–32. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Odnokoz, O.V.; Pavlov, S.V.; Belenicheva, O.I.; Polyakova, E.N. The neuroprotective activity of tamoxifen and tibolone during glutathione depletion in vitro. Neurochem. J. 2012, 6, 202–212. [Google Scholar] [CrossRef]
- El Ouaamari, Y.; Bos, J.V.D.; Willekens, B.; Cools, N.; Wens, I. Neurotrophic Factors as Regenerative Therapy for Neurodegenerative Diseases: Current Status, Challenges and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 3866. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Liu, Y.; Dai, C.-L.; Baazaoui, N.; Tung, Y.C.; Liu, F.; Iqbal, K. Neurotrophic Treatment Initiated During Early Postnatal Development Prevents the Alzheimer-Like Behavior and Synaptic Dysfunction. J. Alzheimer's Dis. 2021, 82, 631–646. [Google Scholar] [CrossRef]
- Clarke, I.J.; Smith, J.T.; Caraty, A.; Goodman, R.L.; Lehman, M.N. Kisspeptin and seasonality in sheep. Peptides 2008, 30, 154–163. [Google Scholar] [CrossRef]
- Masliah, E.; Diez-Tejedor, E. The pharmacology of neurotrophic treatment with Cerebrolysin: brain protection and repair to counteract pathologies of acute and chronic neurological disorders. Drugs Today 2012, 48, 3–24. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Pavlov, S.V.; Dunaev, V.V. Neuroprotective Effect of Cerebrocurin in a Model of Acute Cerebral Stroke. Eksp. Klin. Farmakol. 2010, 73(2):6–9.
- Belenichev, I.F.; Aliyeva, O.G.; Bukhtiyarova, N.V.; Popazova, O.O.; Ryzhenko, V.P. Positive Pharmacological Modulation of Hsp70 in Recovery of Brain Energy Metabolism in Various Models of Cerebral Ischemia. International Electronic Conference on Biomolecules. LOCATION OF CONFERENCE, COUNTRYDATE OF CONFERENCE; p. 24.
- Morguntsova, S.A. Neuroprotection Neurotrophic Cerebroprotector Cerebrocurin in Terms of Modeling Acute Stroke. Zaporozhye Med. J. 2014, 16(5).
- Yevtushenko, O.; Yanovskaya, N.; Yevtushenko, S.; Kutyakova, Y.; Omelyanenko, A.; Dubina, S.; Sokhan, D.; Poroshina, Y.; Fomichova, E.; Sachinova, I. 15-Year Experience of Cerebrocurin Use in Combined Therapy in Children with Organic Diseases of the Nervous System. Int. Neurol. J. 2014, (3.65):13–19.
- Belenichev, I.F.; Sokolik, E.P.; Bukhtiarova, N.V.; Levich, S.V. Pharmacological Modulation of Heat Shock Protein 70 (HSP70)—Dependent Mechanisms of Endogenous Neuroprotection in Conditions of Prenatal Chronic Alcoholism by Cerebrocurin and Tiocetam. 2016, 26, 103–108. [CrossRef]
- Repchuk, Y.; Sydorchuk, L.P.; Sydorchuk, A.R.; Fedonyuk, L.Y.; Kamyshnyi, O.; Korovenkova, O.; Plehutsa, I.M.; Dzhuryak, V.S.; Myshkovskii, Y.M.; Iftoda, O.M.; et al. Linkage of blood pressure, obesity and diabetes mellitus with angiotensinogen gene (AGT 704T>C/rs699) polymorphism in hypertensive patients. Bratisl. Lek. Listy 2021, 122, 715–720. [Google Scholar] [CrossRef]
- Kamyshna, I.I.; Pavlovych, L.B.; Maslyanko, V.A.; Kamyshnyi, A.M. Analysis of the transcriptional activity of genes of neuropeptides and their receptors in the blood of patients with thyroid pathology. J. Med. Life 2021, 14, 243–249. [Google Scholar] [CrossRef]
- Lyubomirskaya, E.S.; Kamyshnyi, A.M.; Krut, Y.Y.; A Smiianov, V.; Fedoniuk, L.Y.; Romanyuk, L.B.; Kravets, N.Y.; Mochulska, O.M. SNPs and transcriptional activity of genes of innate and adaptive immunity at the maternal-fetal interface in woman with preterm labour, associated with preterm premature rupture of membranes. Wiad Lek 2020, 73, 25–30. [Google Scholar]
- Halabitska, I.; Petakh, P.; Kamyshna, I.; Oksenych, V.; Kainov, D.E.; Kamyshnyi, O. The interplay of gut microbiota, obesity, and depression: insights and interventions. Cell. Mol. Life Sci. 2024, 81, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Topol, I.; Kamyshny, A. Study of expression of TLR2, TLR4 and transckription factor NF-kB structures of galt of rats in the conditions of the chronic social stress and modulation of structure of intestinal microflora. Georgian Med. News 2013, 225, 115–122. [Google Scholar]
- Dzhuryak, V.; Sydorchuk, L.; Sydorchuk, A.; Kamyshnyi, O.; Kshanovska, A.; Levytska, S.; Knut, R.; Sheremet, M.; Ivashchuk, S.; Petrynych, O.; et al. The cytochrome 11B2 aldosterone synthase gene CYP11B2 (RS1799998) polymorphism associates with chronic kidney disease in hypertensive patients. Biointerface Res. Appl. Chem. 2020, 10, 5406–5411. [Google Scholar] [CrossRef]
- Halabitska, I.; Oksenych, V.; Kamyshnyi, O. Exploring the Efficacy of Alpha-Lipoic Acid in Comorbid Osteoarthritis and Type 2 Diabetes Mellitus. Nutrients 2024, 16, 3349. [Google Scholar] [CrossRef]
- Nosulenko, I.S.; Voskoboynik, O.Y.; Berest, G.G.; Safronyuk, S.L.; Kovalenko, S.I.; Kamyshnyi, O.M.; Polishchuk, N.M.; Sinyak, R.S.; Katsev, A.V. Synthesis and Antimicrobial Activity of 6-Thioxo-6,7-dihydro-2H-[1,2,4]triazino[2,3-c]quinazolin-2-one Derivatives. Sci. Pharm. 2014, 82, 483–500. [Google Scholar] [CrossRef]
- Bilous II, Pavlovych LL, Kamyshnyi AM. Primary hypothyroidism and autoimmune thyroiditis alter the transcriptional activity of genes regulating neurogenesis in the blood of patients. Endocr Regul. 2021; 55(1): 5-15.
- Halabitska, I.; Babinets, L.; Oksenych, V.; Kamyshnyi, O. Diabetes and Osteoarthritis: Exploring the Interactions and Therapeutic Implications of Insulin, Metformin, and GLP-1-Based Interventions. Biomedicines 2024, 12, 1630. [Google Scholar] [CrossRef]
- Bilyi AK, Antypenko LM, Ivchuk VV, Kamyshnyi OM, Polishchuk NM, Kovalenko SI. 2-Heteroaryl-[1,2,4]triazolo[1,5-c]quinazoline-5(6 H)-thiones and Their S-Substituted Derivatives: Synthesis, Spectroscopic Data, and Biological Activity. Chempluschem. 2015; 80(6):980-989.
Figure 1.
Diseases associated with mitochondrial dysfunction in cellular energy metabolism. The Figure was designed using BioRender.com.
Figure 1.
Diseases associated with mitochondrial dysfunction in cellular energy metabolism. The Figure was designed using BioRender.com.
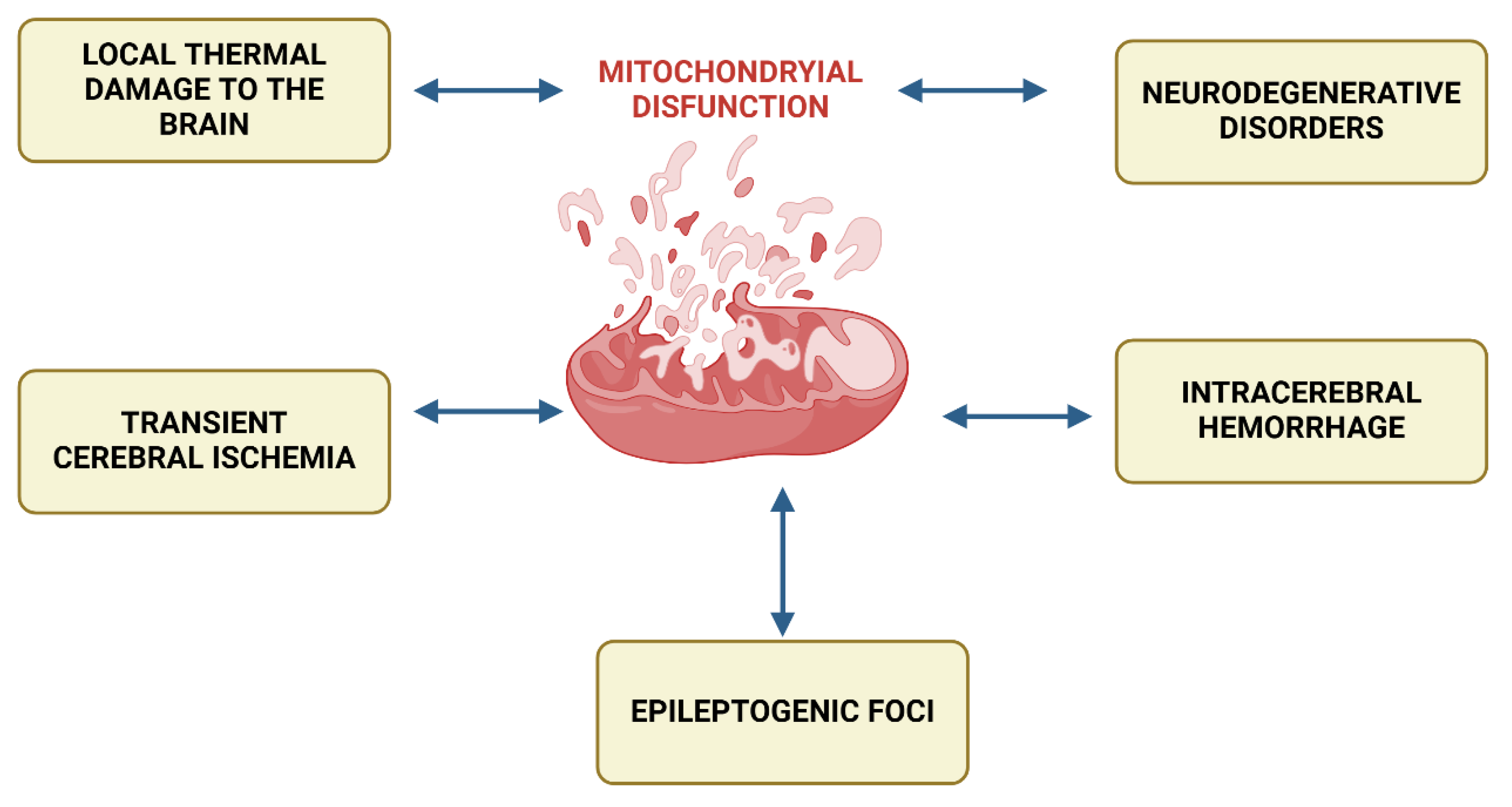
Figure 2.
Diseases associated with primary mitochondrial dysfunction. The Figure was designed using BioRender.com.
Figure 2.
Diseases associated with primary mitochondrial dysfunction. The Figure was designed using BioRender.com.
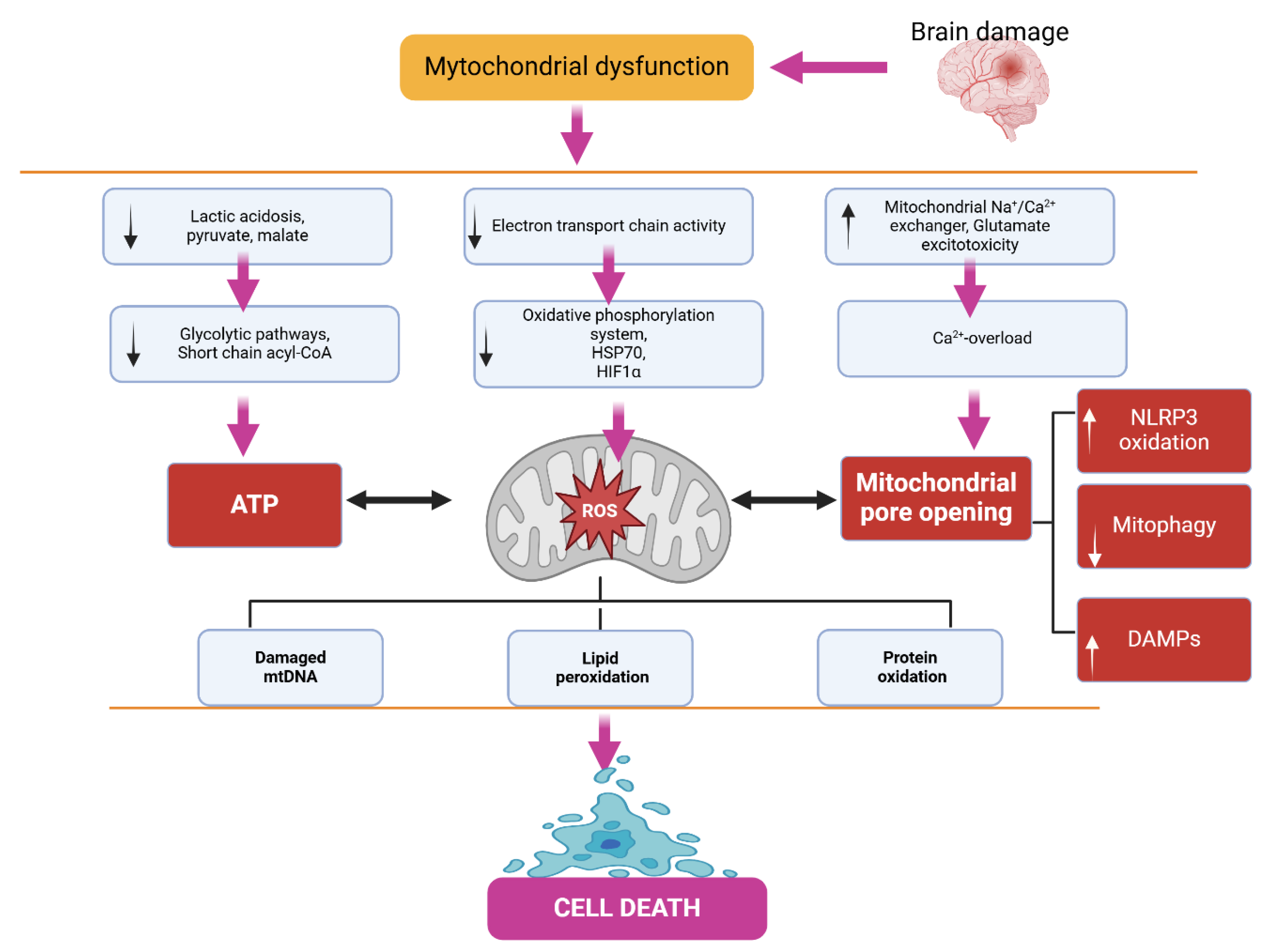
Figure 3.
Disruptions in mitochondrial function. The Figure was designed using BioRender.com.
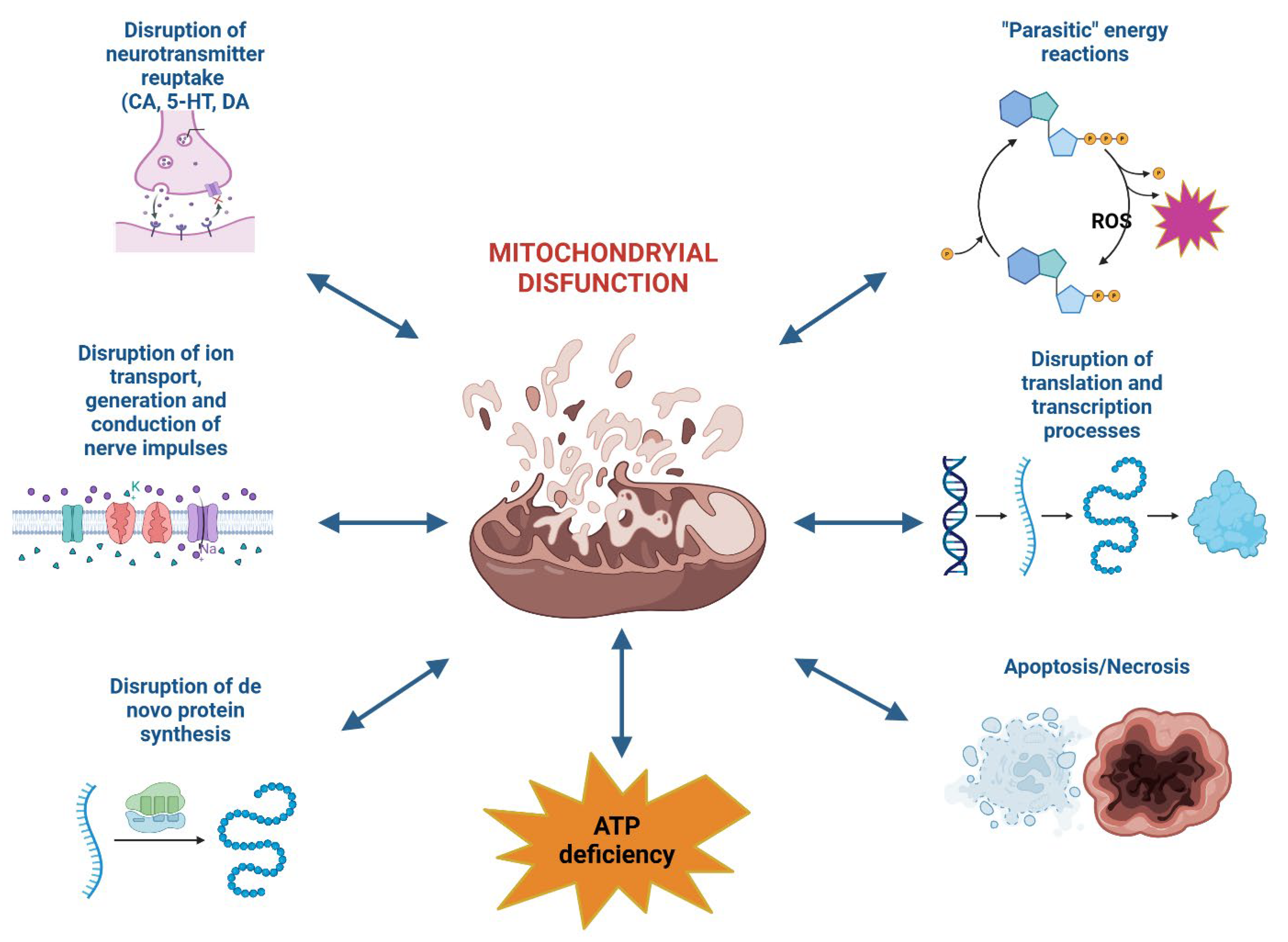
Figure 4.
Action mechanism of Thiotriazoline. The Figure was designed using BioRender.com.
Figure 5.
Action mechanism of Angiolin. The Figure was designed using BioRender.com.
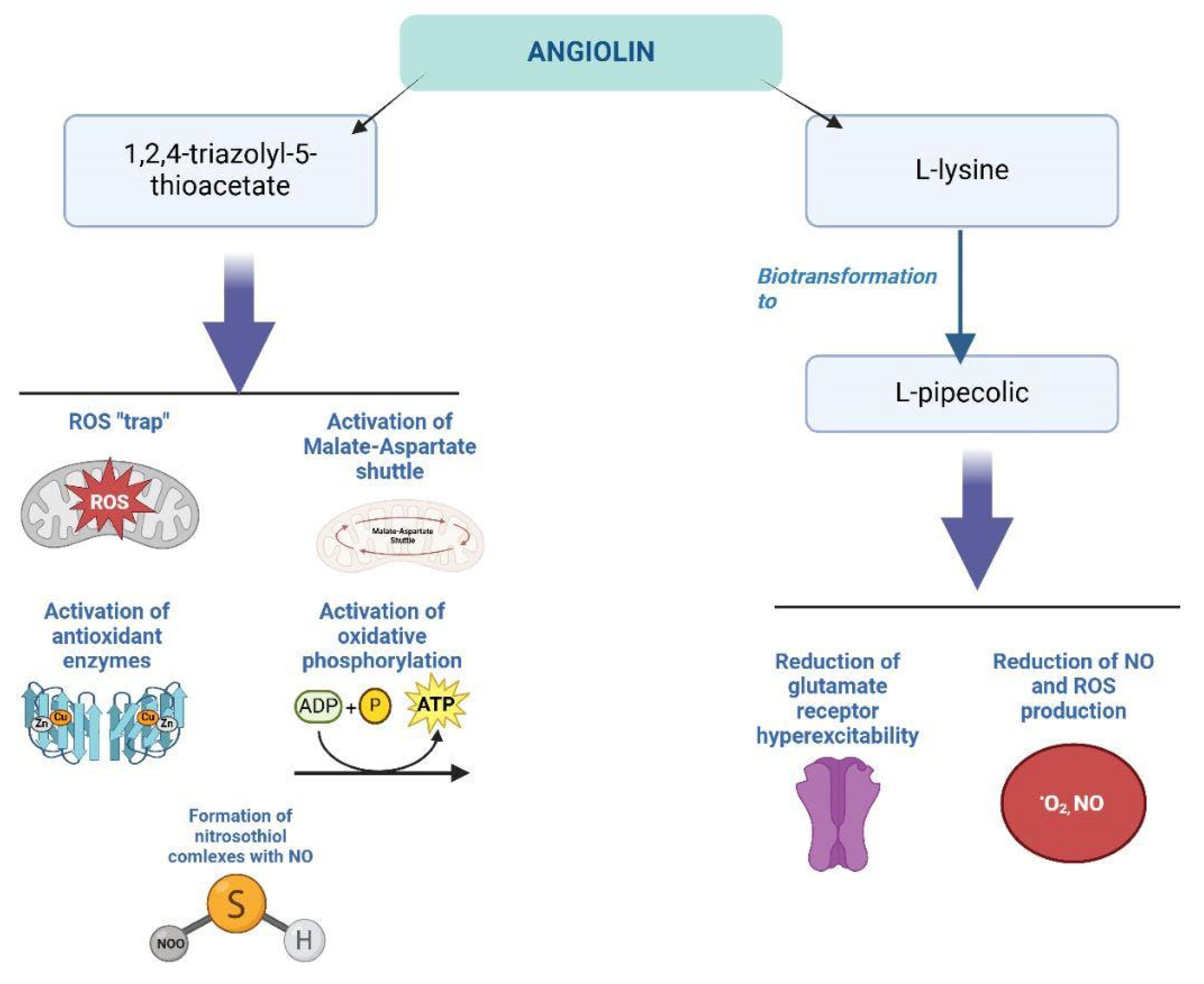
Figure 6.
Action mechanism of Cerebrocurin. The Figure was designed using BioRender.com.
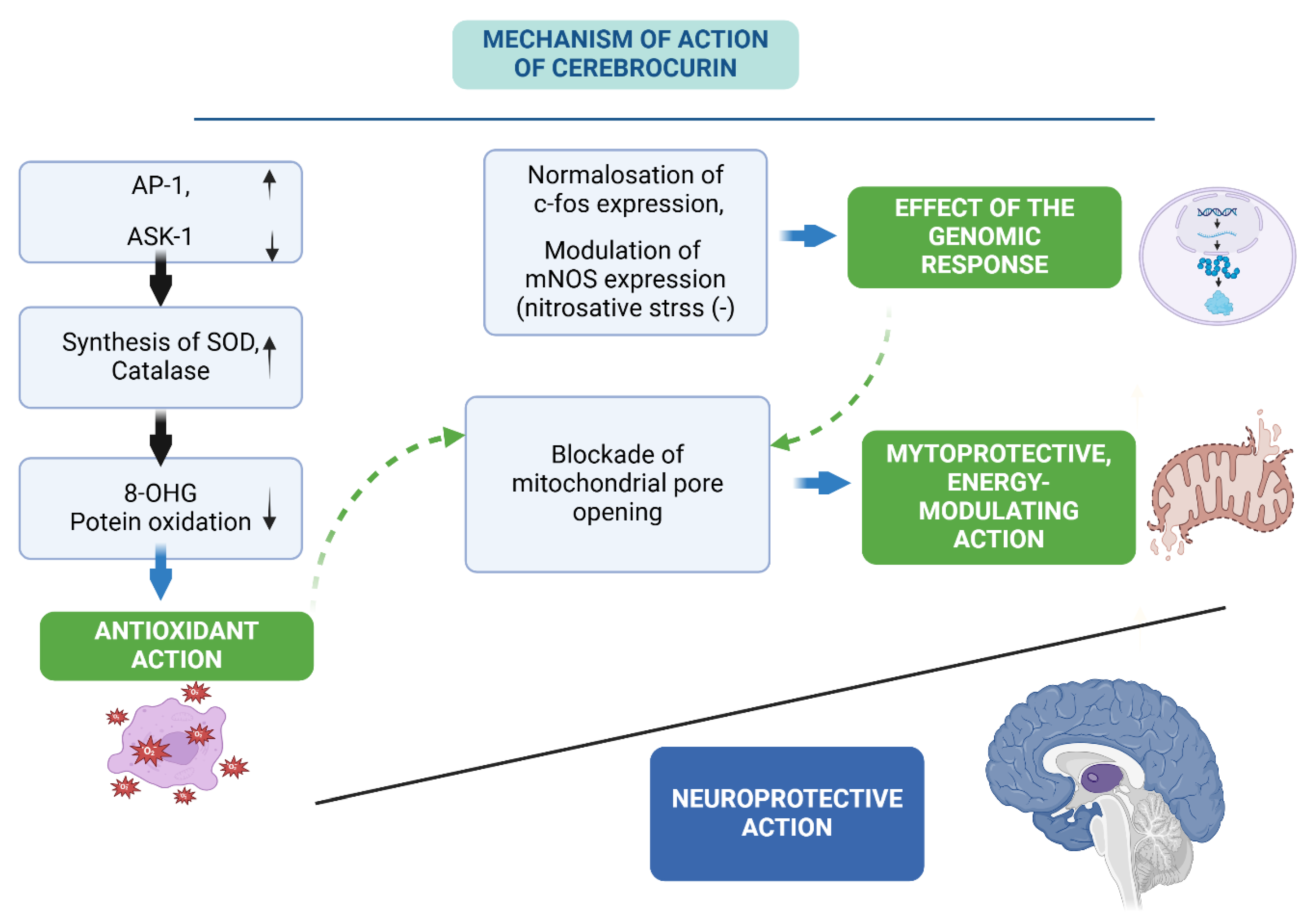
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



