
Submitted:
27 February 2025
Posted:
03 March 2025
You are already at the latest version
Abstract
Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) is a major viral threat to swine, causing significant economic loss in global pig farming industry. The virus exists in two major genotypes, both characterized by high mutation rates and genetic variability, complicating the development of a universally effective vaccine and the control of disease consequently. To address this challenge, a novel multi-epitope-based vaccine was designed to elicit a robust and cross-protective immune response against multiple PRRSV strains. The complete sequences of PRRSV encoded proteins were retrieved worldwide, and the conserved fragments were identified through alignment of polypeptide sequences. Subsequent screening was conducted screened epitopes for their potential to be safe and to activate B cells, CTLs and HTLs. By conjugating the selected epitopes with distinct adjuvant proteins, three vaccine candidates were constructed and termed as PRRSV-Vaccine (PRRSV-V-1, PRRSV-V-2, and PRRSV-V-3 respectively). Furthermore, systematic evaluations of their physiochemistry properties, structural stability, binding with pattern recognition receptors and induction of host immune system were performed. PRRSV-V-2 was demonstrated with most promising physicochemical and structural characteristics, strong binding with TLR3 and TLR8, and most vigorous reactions of host immune response. As the most promising candidate, the recombinant PRRSV plasmid was in-silico designed for expression in Escherichia coli. Our study proposed a novel approach to PRRSV vaccine development against PRRSV, offering a promising strategy for controlling the infection across diverse PRRSV virus strains in swine.
Keywords:
Reverse Vaccinology
; Porcine Reproductive and Respiratory Syndrome Virus
; PRRSV-Vaccine
; In-silico
; multi-epitopes
1. Introduction
Porcine reproductive and respiratory syndrome (PRRS), caused by the PRRS virus (PRRSV), was first identified in United States in 1987. Since then, PRRSV has become a global threat, inflicting severe economic losses in swine industry [1]. Recent estimates show that PRRSV costs pork producers approximately $1.2 billion annually [2,3]. In Germany, financial losses due to PRRSV resulted in a 19.1% average reduction in farm profit, reaching up to 41% under severe conditions in 2021 [4]. One of the key challenges in controlling PRRSV lies in its genetic and antigenic variability [5,6,7], which complicates both disease tracking and development of an effective, universal vaccine. The virus’s high mutations rate has hindered the creation of a broadly protective vaccine, prolonging the fight against this devastating pathogen [8,9].
PRRSV is a member of Arterivirus genus within the Arteriviridae family, with a genome of approximately 15.4 kb and encodes 10 open reading frames (ORFs) [10]. This virus produces two large polyproteins from ORFs 1a and 1b, which are cleaved into 14 nonstructural proteins [11]. Additionally, 7 structural proteins are encoded by ORFs 2–7, including GP2a, GP3, GP4, GP5, E protein, M protein and N protein [12]. These structural proteins play key roles in virus-host interactions and are highly immunogenic, making them key targets for vaccine development. Currently, 365 distinct PRRSV strains have been isolated worldwide, with significant genetic diversity due to ongoing recombination and mutations in the viral genome. Notably, the GP2, GP3, GP4, and GP5 proteins are hotspots for mutations, while the GP6 (M protein) and GP7 (N Protein) sequences are relatively conserved. Based on these variations, PRRSV is classified into two major genotypes: Type I (European) and Type II (North American) [13,14]. The emergence of new strains continues to challenge the development of vaccine capable of providing broad protection across diverse genotypes.
In response to this constant evolution, a variety of vaccine types have been developed, including 17 modified live virus (MLV) vaccines, 3 killed virus (KV), 5 adenovirus-vectored, 2 synthetic peptide, and 14 subunit vaccines, as of 2024 [15,16]. Although MLV and KV vaccines have shown practical effectiveness, each has significant limitations (Supplementary Materials), MLV vaccines are effective against homologous strains but pose safety concerns, such as the risk of virulent reversion and recombination with filed strains [17,18,19]. Currently, a total of five MLV vaccines are used in the U.S., seven in Europe, and four in Asia (Supplementary Materials). However, significant concerns of safety are always accompanied due to the possibility of virulence reversion and recombination with virulent field strains [19,20]. On the contrary, KV vaccines are valued for safety and no adverse effects reported [21], which is benefitted from the killed nature and consequently eliminated risks of replication, mutation, or spreading among vaccinated animals. However, it’s regrettable that KV vaccines are often considered less effective than MLV vaccines [17,19], which limited their market capacity (Supplementary Materials). As reported, enhancement of humoral immune response was constantly disabled with the variety of virus concentration and inactivation methods [22]. Since then, KV vaccines are not yet approved in the United States, despite being approved in Europe and other regions.
To overcome these challenges, identifying and targeting protective epitopes is crucial for developing a more effective vaccine. Several neutralizing epitopes have been identified in PRRSV structural proteins, particularly GP3, GP4, GP5, and M [23,24]. GP5, as a major envelope glycoprotein, plays a key role in viral entry and assembly [25], with neutralizing epitopes found in its N-terminus ectodomain [25,26]. Similarly, epitopes in the M proteins have been identified as important for susceptibility to neutralization antibodies [24,27,28]. In M protein, the patterner of GP5, a fragment of 70-amino acid residues was determine as the crucial modulator of the susceptibility to neutralization antibody in porcine serum [29]. However, partial protection has been observed with immunization using GP5-M ectodomain, suggesting that additional epitopes are needed for full viral neutralization. Alternatively, neutralizing epitopes have also shown in GP2, 3 and 4 [30,31,32] with which another complex is formed as an essential player in receptor binding and viral infection [33,34]. With progression of epitope discovery, new candidates were continuously prompted. For instance, aa37-45 in GP5 of both genotype [27] and aa57–68 in GP4 of type I [32,35], have been reported as neutralizing epitopes. T cell epitopes were also highlighted from GP4 and GP5 [36,37]. In N protein, four to five antigenic regions are included [12] and essential to induce protective B- and T-cell responses [38]. Despite these findings, cross-genotype protection remains a major hurdle due to the high mutation rate of PRRSV antigens, which allows for immune evasion and complicates vaccine development. New epitopes must be identified to update the vaccine formulation and address this challenge.
To overcome the limitations, od current vaccine strategies, an in-silico approach is proposed for identifying novel epitopes and designing a multi-epitope vaccine. With current progression of immunoinformatics, identification of epitopes and formulation of vaccine has become feasible and efficacious [39,40]. Under in-silico strategy, vaccine schemes have been achieved successfully to pathogens, like human coronavirus [41], Dengue virus [42], Influenza virus [43] etc. In current study, we conducted the in-silico design to target either type of PRRSV. The effective and non-risky epitopes were screened from the viral protein fragments conserved across the globally isolated strains. Subsequently, the vaccine candidate was constructed through assembly of selected epitopes and diverse adjuvant proteins. With evaluation of candidates in physiochemistry, structure biology and immunology, one promising candidate was highlighted ultimately. This study aims to provide a promising strategy for overcoming the challenges of PRRSV vaccine development, offering cross-genotype protection despite of the genetic diversity of PRRSV.
2. Materials and Methods
As illustrated in Figure 1, the PRRSV vaccine design was carried in stepwise manner. The web links to databases and online platforms used in this study are listed in Supplementary File S1, Table 4.
2.1. Retrieval and Analysis of PRRSV Protein Sequence
To initiate the design, full-length sequences of all nonstructural and structural proteins from both PRRSV genotype were retrieved from NCBI (Supplementary Tables S5 and S6), focusing on sequences deposited between 2018 and 2023. Phylogenetic analysis and evolutionary distance calculations for each viral protein were performed using Molecular Evolutionary Genetics Analysis (MEGA) software, version 4 [44]. Construction of phylogenetic trees was conducted using the neighbor-joining method, and the results were visualized with Interactive Tree Of Life (iTOL) [45]. Conserved fragments within each protein were identified using multiple sequence alignment (MSA). Initial alignment was conducted using MUSCLE [46], followed by confirmation with CLUSTALW version 2.0 [47] and MAFFT [48]. Conserved sequences shared across retrieved strains were extracted using BioEdit version 7.2 [49] and are listed in Supplementary Table S7.
2.2. Step-by-Step Selection of Epitopes
Identification of epitopes for B Cells, CTLs, and HTLs was carried out using IEDB tools based on the conserved protein fragments in each viral protein.
For linear B-cell epitopes, the Bepipred Linear Epitope Prediction 2.0 method was applied, with a minimum epitope length of 6 amino acids [50]. The threshold of 0.5 was set to determine the likelihood of an immunogenic epitope. NetMHCpan EL4.1 was used with “Pigs” specified as the MHC source to predict CTL epitopes. Epitope lengths were defined as 9 and 10 amino acids, targeting the most common SLA class I alleles [51]: SLA-1*0702, SLA-1*1101, SLA-2*0101, SLA-2*1101, SLA-2*1201, SLA-3*0101, SLA-3*0401, SLA-3*0501, SLA-3*0701. Only peptides ranked within the top 1% of affinity were considered for further analysis. The prediction of HTL epitopes was conducted with NetMHCIIPan 4.1 EL. Since there was no SLA options for MHCII prediction, the HLA reference was set as below: HLA-DPA1*02:01/DPB1*05:01, HLA-DRB4*01:04, HLA-DRB1*12:04, HLA-DPA1*02:01/DPB1*04:01, HLA-DRB5*01:03, HLA-DPA1*01:03/DPB1*01:01, HLA-DPA1*02:01/DPB1*02:01, HLA-DRB1*04:04) [52]. A fixed length of 15aa was used in the prediction, and the top 0.5% of affinity was selected for further evaluation.
Selected epitopes for all three types were assessed for conservancy, antigenicity, allergenicity, and toxicity with IEDB analysis resource [53], VaxiJen v2.0 [54], AllerTOP v.2.0 [55] and ToxinPred [56], respectively. The inclusion criteria required sequence identity ≥80%, antigenicity score >0.5 and non-allergic, non-toxic properties. Additional scoring of immunogenicity was conducted for CTL epitopes with Class I Immunogenicity server [53] and the ones scored ≤0.2 were excluded. For HTL epitopes, further prediction were made to identify potential inducer of IFN-γ and IL-4 using the IFNepitope [57] and IL4pred [58] servers.
2.3. Design and Assembly of Candidate Vaccines
The candidate epitopes that passed the stepwise selection process were then used for vaccine construction. Initially, the epitopes were categorized based on their reactivity with B cells, CTLs, and HTLs, and subsequently assembled into a core antigen. Distinct adjuvant domains were attached to enhance the immune response, as shown below:
S50 L7/12 ribosomal protein, isolated from mycobacterium tuberculosis, known for the ability to provoke an immune response [59].
β-defensin, recognized for its antimicrobial and immunomodulatory properties [60].
Heparin-binding Hemagglutinin (HBHA), sourced from Mycobacterium species, known for its immunogenic properties [61].
For vaccine assembly, and further reinforcement of immunogenicity, the following linkers were used to organize the immune elements [59]:
KK: linker to connect B-cell epitopes [62].
AAY: linker used for organization of CTL epitopes [63].
GPGPG: linker to group HTL epitope [42].
EAAAK: a rigid linker to fuse the adjuvant domains to the N-terminus of the core antigen.
2.4. Evaluation of Candidate Vaccines in Physicochemical and Immune Properties
The ExPASy program [64] was used to predict a range of physicochemical properties for the candidate vaccines, including molecular weight, isoelectric point, half-life, stability, and other essential characteristics. Solubility of these molecules were predicted using SOLpro [65].
Additionally, the immune properties of the candidate’s vaccine were assessed using immunoinformatic tools, including VaxiJen v2.0 for immunogenicity scoring, ANTIGENpro for antigenicity, AllerTop for allergenicity assessment, and ToxinPred to evaluate toxicity.
2.5. Prediction and Validation of Molecular Structure for Candidate Vaccines
The secondary structure of the vaccine molecules, including α-helices, β-strands, and coils, was predicted using PSIPRED [66] based on their polypeptide sequences.
2.6. Molecular Docking and Dynamic Analysis of Vaccine-TLR Complexes
To evaluate the potential recognition of vaccine molecules by host immune system, molecular docking was performed between the candidate vaccines and two pattern recognition receptors, TLR3 and TLR8 of swine. The tertiary structural of receptors was retrieved from the RCSB protein data bank [71,72] with PDB IDs D0V2A2 for TLR3 and Q865R7 for TLR8. Docking was conducted using the HDOCK server, and 10 resulting docking models were returned, ranked by lowest binding energy [73]. For each vaccine-TLR complex, model 1# with the lowest binding energy was selected for analysis of non-covalent interactions, including hydrogen bonds, salt bridges, and interface areas, using the PDBsum [74] server. The 3D structures with related interactions were then visualized by PyMOL [75].
For the top-ranked PRRSV-V-2 -TLR complexes, Molecular Dynamics (MD) simulations were performed using the AMBER18 platform [76]. A 14-angstrom padding around the protein and water box were applied. The FF14SB force field [77,78] was used for both vaccines and TLRs. The vaccine molecules were placed in a TIP3P water box with counterions added to neutralize the system. To minimize structural collisions, steepest descent or conjugate gradient minimization methods were employed, with transition steps every 1000 cycles up to 5000 cycles. Thermal equilibrium was reached at 300 K after 250 picoseconds (ps) of system heating. A production run of 100 nanoseconds (ns) was conducted to explore the dynamic behavior of the vaccine–receptor complexes. In the canonical ensemble, periodic boundary conditions were applied to maintain the rigidity of the covalently bound hydrogen atoms using the SHAKE algorithm [79]. In these simulations, the temperature was regulated at 300 K using a Langevin thermostat with a 10 Å cutoff for non-bounded interactions and Ewald simulations for long-range interactions. Statistical parameters such as the root mean square deviation (RMSD), root mean square fluctuations (RMSF), and radius of gyration (Rg) of vaccine-receptor complexes were analyzed using the CPPTRAJ module [80]. The results were visualized in PyMOL [81] to assess the structural stability and dynamic properties of the vaccine-receptor.
2.7. Immune Simulation of Candidate Vaccines
Immune simulations were conducted using the C-ImmSim server [82] to model the immune responses elicited after vaccines administration according to a specified immunization schedule. The simulation protocol involved three doses of immunization with two-week interval, corresponding to simulation steps 1, 84, and 168, where each step represented eight hours of real-time. Default settings were used for all additional parameters to ensure consistency and standardization.
2.8. In Silico Cloning of Selected Vaccine Candidate
To generate the DNA coding sequence of vaccine protein, JCat server were used for reverse translation and codon optimization based on E. coli codon usage bias. The codon adaptation index (CAI) and GC content percentage of the resulting sequence were calculated to assess protein expression level [83]. A good outcome was defined as a CAI > 0.8 while GC content ranging 30–70%. Furthermore, in-silico cloning was performed in SnapGene using the pET-28a (+) vector, with restriction sites Xhol and BamHI.
3. Results
3.1. Extraction of Conserved Fragments from PRRSV Protein Sequences Collected Worldwide
Due to the nature of rapid evolution, PRRSV protein sequences deposited between 2018~2023 were taken as the starting material for current design. 7315 full-length sequences were downloaded in total from NCBI in FASTA format, including 90 proteins for PRRSV1 and 7225 for PRRSV2. Protein accession numbers were listed in Table S4. As showed in phylogenetic analysis, sequences of same proteins were tightly grouped in the trees (Figure S1). Based on the alignment, amino acid sequences among the retrieved sequences identified conserved regions in both PRRSV. Based on the alignments, conserved fragments were identified for each strain, and those with at least 10 amino acids were used for epitope screening (Table S5). Finally, 30 conserved fragments were obtained for PRRSV-1, and 77 for PRRSV2.
3.2. Prediction and Evaluation of Epitopes for B Cells, CTLs and HTLs
B cell epitopes prediction was performed based on structural proteins of PRRSV, including GP2a, GP3, GP4, GP5, M and N. Primarily, 68 epitopes were generated for Type1 and 97 for Type2. Epitopes were then evaluated for their antigenicity, allergenicity and toxicity. Subsequently, the ones of antigenic, non-allergic and non-toxic were reserved for further usage. Conservancy of epitopes was also analyzed using BCPRED algorithm in IEDB platform and above 50% conserved ones were highlighted from the reserved epitopes. The ultimate selection of B cell epitopes was included in Table 1, the procedure of epitope selection was presented in Figure S2 and Tables S8 and S9.
Epitopes for T-lymphocytes were predicted from all PRRSV proteins, and 4208 epitopes were generated for CTLs and 4560 for HTLs (Figures S3 and S4). The first selection was then carried out based on binding affinity between candidate epitopes and MHC I or II alleles, with top 10% and top 1% as the threshold respectively. Further selection of epitopes was conducted for both CTLs and HTLs to highlight the most promising inducer of T cell reaction (Tables S10 and S11). For CTLs, epitopes were chosen once antigenicity scored >0.5 and immunogenicity >0.1, while excluded if allergenic, toxic or conservancy lower than 50%. Similar filters were also set up for HTLs, but capacity of IFN-γ and IL-4 induction was evaluated instead of immunogenicity scoring (Tables S12 and S13). Resultantly, 10 HTL epitopes and 10 CTL epitopes were elected for final vaccine design (Table 2 and Table 3).
3.3. Construction of Three Candidate Vaccines
To construct the candidate vaccines, core antigen was firstly organized via assembly of selected epitopes of B-cells, CTLs and HTLs with help of linker sequences. KK, GPGPG and AAY peptides were employed to group three classes of epitopes (Figure 2A). after that, three distinct adjuvant proteins, L7/12 S50 ribosomal protein, β-defensin and HBHA, were adopted to enhance the boosting of host immune system. The adjuvant proteins were fused with core antigen at N-terminus with EAAAK, a rigid linker. Correspondingly, three candidate vaccines were generated and termed PRRSV-V-1, PRRSV-V-2 and PRRSV-V-3 as shown in (Figure 2B–D), while protein sequences of vaccine candidates were contained in Figure S5:
PRRSV-V-1: Assembling the core Antigen with S50 L7/12 ribosomal protein on N terminal PRRSV-V-2: Core Antigen attached with β-defensin on the N-terminal PRRSV-V-3: HBHA adjuvant assembled with core antigen on N terminal
3.4. Properties of Candidate Vaccines in Immunology and Physiochemistry
To assure the qualification of candidate vaccines primarily, their antigenicity, allergenicity and toxicity were analyzed before any other characteristics (Table 4). All three molecules were showed non-allergic, non-toxic, and scored >0.6 in antigenicity (line 9-11 in Table 4), which is demanded basically to be safe and effective in immunization. The physicochemical properties were analyzed thereafter to assess their molecular size, stability, solubility, and hydropathicity, which were correlated with the feasibility in the future preparation and application of candidate vaccines. For the molecular size, all three candidate vaccines PRRSV-V-1~3 contained approximately 531Amino Acids,448Amino Acids, and 551 amino acids (line1in Table 4) and weighted around 50KDa,49KDa, and 60KDa (line 2 in Table 4). To instruct a good stability in vitro, Instability Indexes were calculated much lower than 40(line 3 in Table 4). For the intra-cellular stability, the protein half-life was estimated more than 30 hours in mammalian cells, beyond 20 hours in yeasts, and in E. coli no less than 10 hours (line 5 in Table 4). The water solubility of candidates was scored with SOLpro, and in addition, by ProtParam server. Resultantly, the solubility scores were around 0.9(line 6 in Table 4), pI generally above 9(line 8 in Table 4), Aliphatic Index ranging 69~82(line 4 in Table 4), GRAVY slightly below 0(line 7 in Table 4). In one word, these three candidate vaccines were suggested as hydrophilic and well soluble in water of physiological pH.
3.5. Prediction and Refinement of Secondary and Tertiary Structure
To determine the composition of secondary structural elements in candidate vaccines, Psipred server was used. In PRRSV-V-1, there are 44.06% of alpha helix, 11.11% of beta-strand and 44.82% of random coil, as plotted along its polypeptide sequence. The alpha helix, beta-strand, and random coil in PRRSV-V-2 were 43.30%, 12.05% and 44.64%, while 57.16%, 6.71% and 36.11% in PRRSV-V-3, respectively (Figure 3A and Figure S6).
For the modeling of tertiary structure, templates c8yeqA, c1kj6A and c2ch7B were employed on Phyre2 platform in corresponding to PRRSV-V-1, 2 and 3 candidates (Figure S7). The levels of confidence and coverage were then showed as Figure 3B,C. The structural models were then verified with Procheck and the Ramachandran plots generated subsequently. As visualized in Ramachandran plot, 96.7% residues in PRRSV-V-1 were located into the favored region, 3.3% in allowed region and 0% in disallowed region. Similarly, the residues of PRRSV-V-2 and 3 were mainly in favored region (Figure 3D and Figure S8). On the other side, ProSA-web showed Z-scores as -5.98, -4.80 and -1.24 for vaccine candidates PRRSV-V-1, 2 and 3, respectively, with which high quality was indicated for all three models (Figure 3E and Figure S9).
3.6. Molecular Docking and Dynamics Simulation for Complexes of Vaccine-Immune Receptor
To evaluate the interaction between vaccine molecule and patterning recognition receptors, molecular docking was performed using HDock server. Two receptor molecules of swine, TLR3 and TLR8, were used in docking with three candidate vaccines (Table S14), and then 6 complexes were generated. For each complex, 10 models were constructed and ranked by docking score from low to high, among which the model 1# exhibited the possible strongest binding.
Tight binding between vaccines and receptors was suggested with lowest binding energy scored ranging -100~-350 kJ/mol (Table S15). The interactions were illustrated in detail, while the number of hydrogen bonds, salt bridges, non-ionic interactions and size of interface were listed in Figure 4A. To PRRSV-V-1, complexes with TLR3 and 8 were scored as -196.74 and -190.43 kJ/mol, respectively. The interface between PRRSV-V-1 and TLR3 was composed with 15 and 20 residues in vaccine and receptor, respectively. Area of this interface reached 1104 Å2, within which 3 hydrogen bonds, 2 salt bridges, and 121 non-covalent interactions were formed. An interface of much smaller scale was formed between PRRSV-V-1 and TLR8, which contained much fewer numbers of connections, including 2 hydrogen bonds, 0 salt bridges, and 87 non-covalent interactions. To the other 4 complexes formed with PRRSV-V-2 and -3, scores lower than -260 kJ/mol were determined in docking. Much higher number of hydrogen bonds and non-covalent interactions were formed between PRRSV-V-2 and TLR3 or TLR8. Regarding PRRSV-V-3, interfaces and non-covalent interactions were established with either receptor in a dramatically higher scale. Suggested by better docking score and tighter interactions, PRRSV-V-2 and -3, but not PRRSV-V-1, was preferred to be captured and recognized by the host immune system.
Simulation of molecular dynamics was further carried out to substantiate the conformational stability of vaccine-receptor complexes. RMSD was quantified to indicate the robustness of vaccine-receptor complexes, and Rg for compactness. For PRRSV-V-2, RMSD value was restrained under 4 (Figure 4B) while Rg value ranged 31.5~33.5 with either TLR3 or TLR8 (Figure 4C). The hydrogen bonds were also formed constantly between 8 to 16 inside the complexes (Figure 4D). However, regarding PRRSV-V-1 or -3 (Figures S11 and S12), RMSD values were roughly above 5, even up to 30 when PRRSV-V-1 coupled with TLR3. On the other hand, Rg level was fluctuated from around 34 to 55 for either vaccine candidate. Thus, it can be seen, stable complexes can be formed by PRRSV-V-2 with TLR3 or TLR8, rather than PRRSV-V-1 or -3. Accordingly, PRRSV-V-2 will be subjected to further simulation of immune.
3.9. In Silico Stimulation of Immune Responses
To demonstrate the efficacy of vaccine candidate PRRSV-V-2 to induce host immune, simulation was then conducted on the platform C-ImmSim with three doses of vaccination by 4 weeks interval till 350 days post immunization. To see the status of innate immunity, dendritic cells, macrophage cells and nature killer cells were monitored in simulation. A significant activation of macrophage cells was demonstrated following each dose of immunizations. In addition, the level of cytokines like IFN-γ and IL-2 were also elevated rapidly up to 400,000 and 700000 ng/ml, respectively. Thus, it can be seen that vaccine candidate PRRSV-V-2 is able to initiate the innate part of host immune system shown in Figure 5.
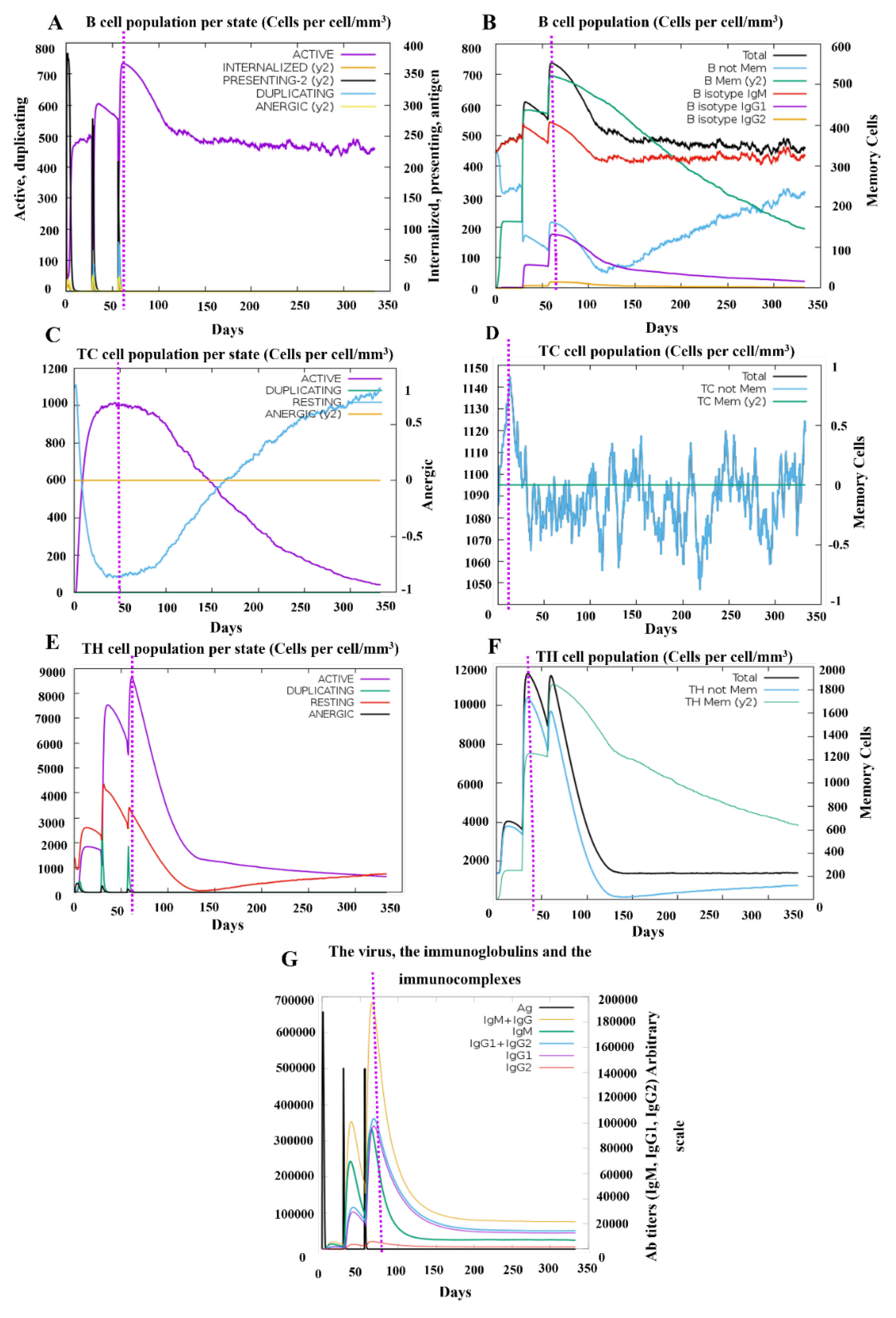
The adaptive immune response was also analyzed in this simulation to assess the activation of B cells, T cells and immunoglobulins, with a particular focus on the B and T cell related immune memory.
Following three vaccine doses, a robust and long-lasting activation of B cells was observed, with a notable increases in memory B cells and IgM isotypic B cells, which were persisted till one year after the initial immunization. Production of antibodies was observed in a pattern following the dynamics pattern corresponding to B cells activation. The total levels of IgM and IgG peaked at nearly 200000, with individual levels reaching around 100000 for each, suggesting a potent humoral response comparable to other published vaccines designed in silico [84]. T cell activation was also evident, with strong responses in both cytotoxic and helper T cell populations. Notably, memory helper T cells displayed high dynamic activity, maintaining a level above 600 at 350 days post-immunization, which corresponded to 30% of the peak response (Figure 7). The peak immune response across different parameters was achieved approximately 50 days after the first dose, coinciding with the period immediately following the third vaccination.
These findings highlights the vaccine’s robust immunogenic potential, suggesting that it can induce strong and systematic adaptive immune protection against PRRSV infection. Especially, prolonged memory B and T cells responses indicate the potential for long-term immunity following immunization.
Figure 6.
Simulation of the adaptive immune response following PRRSV-V-2 vaccination. (A) status of the B-cell population post-vaccination is represented, with subpopulations categorized as active, internalized (y20, presenting-2 (y2), duplicating, and anergic (y2).(B) Distribution of B-cell subpopulations based on the antibody production status, indicating the total population divided not mem, Bmem(y20). (C, D) Dynamics of CD8+ cytotoxic T-cells post-vaccination are shown, with distinct activation statuses (C), Such as active, duplicating, resting, (D)anergic(y2), and memory conditions. (E, F) post-immunization trend CD4+ helper T-cells are displayed, categorized by activation status (E), such as active, duplicating, resting, and (F) energic(y2), and memory conditions (G) Immunoglobulin production following three doses of PRRSV-V-2, showing antigen (Ag) levels. Dash lines were added in the plots to indicated the peak level and corresponding time point for active or total amount of B cells, CTLs, HTLs and immunoglobulin.
Figure 6.
Simulation of the adaptive immune response following PRRSV-V-2 vaccination. (A) status of the B-cell population post-vaccination is represented, with subpopulations categorized as active, internalized (y20, presenting-2 (y2), duplicating, and anergic (y2).(B) Distribution of B-cell subpopulations based on the antibody production status, indicating the total population divided not mem, Bmem(y20). (C, D) Dynamics of CD8+ cytotoxic T-cells post-vaccination are shown, with distinct activation statuses (C), Such as active, duplicating, resting, (D)anergic(y2), and memory conditions. (E, F) post-immunization trend CD4+ helper T-cells are displayed, categorized by activation status (E), such as active, duplicating, resting, and (F) energic(y2), and memory conditions (G) Immunoglobulin production following three doses of PRRSV-V-2, showing antigen (Ag) levels. Dash lines were added in the plots to indicated the peak level and corresponding time point for active or total amount of B cells, CTLs, HTLs and immunoglobulin.
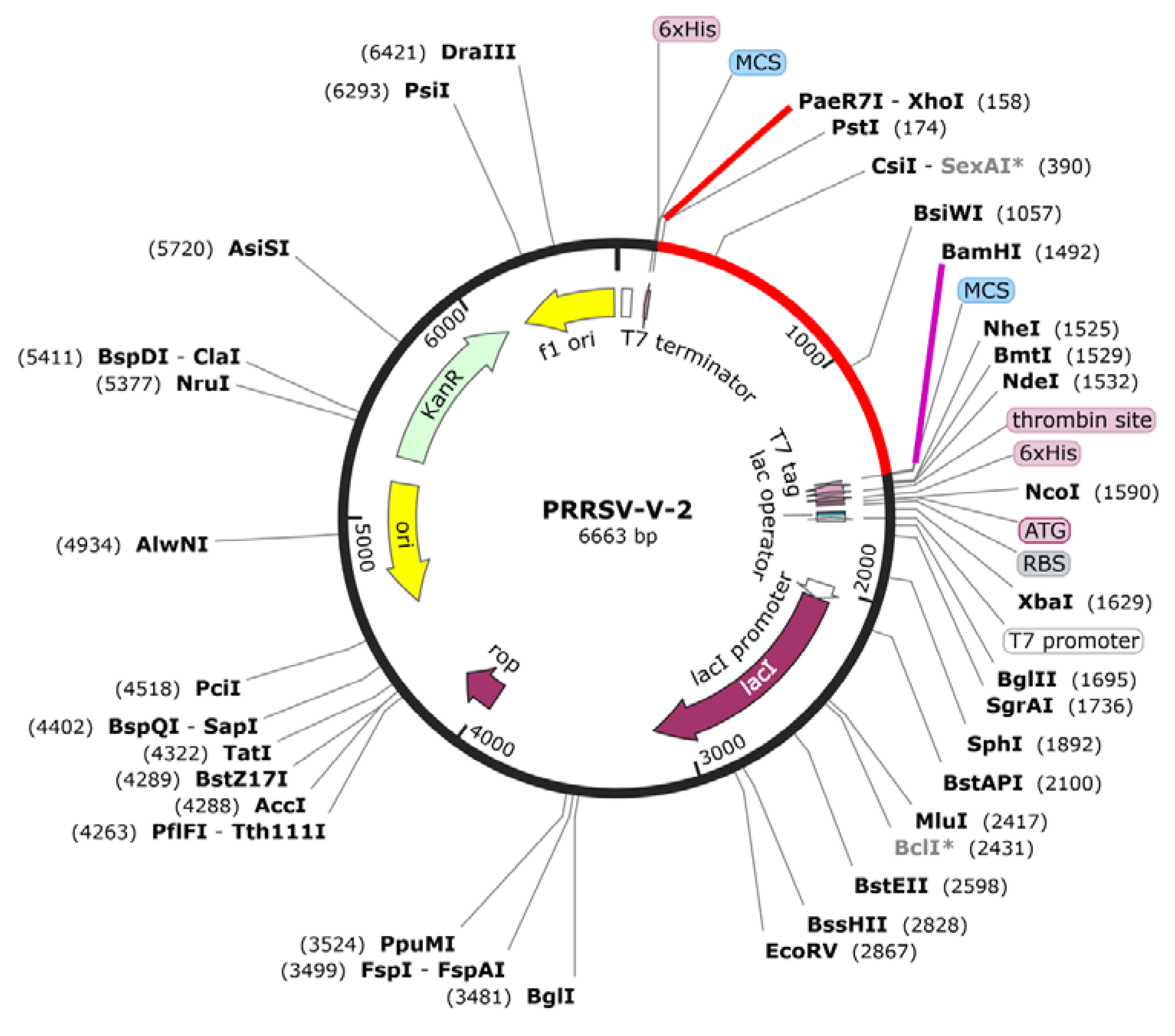
3.10. Codon Optimization of the Final Vaccine Construct
To produce the designed vaccine, its coding sequence was first generated using JCat server. Codon optimization was then performed with JCat to align the codon usage bias of E. coli K12. The optimized DNA sequence, consisting of 1386 bp, had a GC content of 56.02% and 0.90 of CAI, indicating efficient expression potential. This sequence was subsequently, cloned into the pET-28a (+) vector between XhoI and BamHI restriction sites. As a result, a recombinant plasmid for vaccine expression was successfully constructed, ready for application in E. coli expression system.
Figure 7.
A) PRRSV-V-2 codon optimization and plasmid vector construction. The final construct was adapted for use in the pET-28a (+) to form the recombinant plasma sequence of protein expression. The red sequence (gene of interest) is the PRRSV codon sequence optimized for the final designed vaccine. The cloning was done in SnapGene6.1.1.
Figure 7.
A) PRRSV-V-2 codon optimization and plasmid vector construction. The final construct was adapted for use in the pET-28a (+) to form the recombinant plasma sequence of protein expression. The red sequence (gene of interest) is the PRRSV codon sequence optimized for the final designed vaccine. The cloning was done in SnapGene6.1.1.
4. Discussion
Porcine reproductive and respiratory syndrome (PRRS) has been a significant veterinary disease for over three decades, causing substantial economic losses and affecting pig farms worldwide. Despite extensive research, specific therapeutic drugs remain ineffective, and current vaccines offer only limited protection. Moreover, current commercial vaccines offer only modest protection. Even worse, among the technical platforms ever tested, only modified live virus [19] or killed virus [18]-based vaccines have proven effective. These challenges have made the control of PRRS exceptionally difficult.
Several key issues complicate the development of an effective PRRSV vaccine [84]. First, the extensive genetic variations in PRRSV genomes complicated the host immune response, making it difficult to achieve full protection across strains, which only achievable with strains homologous to the vaccine [85]. Second, PRRSV is prone to genetic recombination and replacement, particularly among RNA viruses. This poses a substantial risk of virulence reversal or genetic recombination with field strains when live attenuated vaccines are used [86]. Although inactivated vaccine can mitigate this risk, they do not effectively promote cellular immunity. Furthermore, certain viral proteins interact with the host immune system, evading both innate and adaptive immunity, which either prolongs infection or reduces the immunogenicity of vaccines [87]. Thus, in-depth investigation and continuous development new vaccines with great potency will provide targeted opportunities the prevention and control of PRRSV. However, a long way of struggle is still in demanding to bring forth the vaccine designing.
With the advancement of an in-silico immunoinformatic platforms, a new path for vaccine development has emerged, distinct from the traditional approaches. Instead of relying on a small number of representative strains in wet-lab, large panels of strains from the same species can be utilized virtually to identify epitopes. This approach can enhance the quality and coverage of vaccines. By predicting and scoring epitopes using various bioinformatics tools, promising epitopes can be selected, while suboptimal or risky ones are excluded. Subsequently, This method enables the quick construction of candidate vaccines by combining selected epitopes with adjuvant proteins, followed by multidisciplinary evaluations [88]. Based on the predicted structural and immunogenic properties, the range of optimized proposal is narrowed down from a panel of candidates before conducting actual of wet-lab assays. This approach has facilitated the de novo design of vaccines for a wide range of pathogens, particularly those with significant complexity and challenges in vaccine development. With the benefit brought out with such effective strategy, an earlier design of PRRSV vaccine had been presented by Dr. Dongyu Liu in 2024. However, the study acknowledged certain limitations that need further investigation.
In our study, a multi-epitope vaccine was developed to protect the swine from either PRRSV1 or 2, eliciting both humoral and cellular immune responses. In this vaccine, epitopes were screened from 30 and 77 conserved fragments of viral proteins in Type1 and Type2 PRRSV virus respectively, and selected 6, 10, and 10 epitopes to target B-cells, CTLs, and HTLs. Stringent criteria based on antigenicity, immunogenicity, allergenicity, toxicity, conservancy, and the capacity of cytokine induction were applied. Three candidate vaccines were generated by organizing the selected epitopes and conjugating them with adjuvant protein such as S50 L7/12 ribosomal protein, β-defensin and HBHA. In-silico evaluations demonstrated good structural properties, tight interactions with host pattern recognition receptors, and strong immune stimulation. PRRSV-V-2 emerged as the most suitable candidate due to its favorable interaction with TLR8.
When compared to Dr. Liu’s 2024 in-silico PRRSV vaccine design [71], our approach differed significantly. While Dr. Liu’s design was based on pathogenic isolates specific to his research group, our study included PRRSV protein sequences from global strains, providing a broader representation. Additionally, we used all viral proteins, focusing on conserved fragments across strains, whereas Dr. Liu’s design concentrated on the entire GP3 and GP5 molecules. This difference potentially offers a wider range of epitope sources, improving the vaccine’s ability to cover a broader array of PRRSV strains globally. At the next stage, screening of epitopes was carried in either work, however the evaluations of allergenicity and toxicity were absent in the earlier design by Dr. Liu. In speculation, this was due to the limited supply of source antigen, however, potential risks were then introduced. Unlike that, with the plenty source of viral antigens, enough number of resultant epitopes were still selected in our work, even after stringent evaluation of risks to each single epitopes predicted originally. With the abundance of outcome, more CTL and HTL epitopes were hired to construct the vaccine candidates. Possibly, this will be a positive factor to reinforce the T-cell immunity, which is equivalently important in control of disease and restraining of severity post infection.
In the stages after epitopes obtained, more adjuvant proteins and patterning recognition receptors were tried in our work. Especially, rather than human TLR3 used in Liu’s study, docking of our candidate vaccines was performed with TLR3 and TLR8 of swine, the prospective host. This approach offers more accuracy in predicting the real-world interaction between vaccines and receptors in the host. Additionally, our immune simulation extended to 350 days, compared to just 35 days in Dr. Liu’s study, providing a more comprehensive understanding of immune persistence. With longer observation, the persistence of immune protection was demonstrated in candidates of our selection. The scale of total and active B cell populations was maintained in high level up to one year, while the production of immunoglobulins was also retained in a certain degree. Notably, for the memory B and T cells, about 30% of peak levels were reserved till the end of simulation, which consolidate the confidence to our strategy and choice in current design.
5. Conclusions
In this study, we successfully developed a novel multi-epitope vaccine to address PRRSV’s high mutation rate and genetic variability. We identified key epitopes that effectively induce robust immune responses against B cells, CTLs, and HTLs by using conserved protein fragments from multiple strains. Among the three vaccine candidates evaluated, PRRSV-V-2 demonstrated the best physicochemical properties, strong binding to immune receptors (TLR3 and TLR8), and significant immune system activation. Its recombinant plasmid was designed for expression in E. coli, further validating its potential as a viable vaccine. This research not only presents a promising strategy for developing a broadly protective PRRSV vaccine but also offers a new approach to improving disease control in the swine industry, mitigating the impact of diverse viral strains. However, preclinical and clinical studies are required to validate these findings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary File S1 Tables, Supplementary File S2 Figures.
Author Contributions
Conceptualization, L.T. and S.U.; methodology, S.U.; software, S.U.; validation, L.T.; and S.U., formal analysis, S.U.; investigation, L.T.; resources, H.U. and K.F.; data curation, S.U.; writing—original draft preparation H.U.L.T.; writing—review and editing, L.T.; visualization, L.T.; supervision, L.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research project was funded by “Diagnosis and therapy of viral infectious diseases related to reproduction” and was supported by the startup foundation of new faculty in Nanjing Medical University: Grant number NMUR20240018.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available in the manuscript and Supplementary Materials.
Acknowledgments
This project was supported by the One Hundred Talented Youth Program of the Chinese Academy of Sciences, as well as the startup foundation for new faculty in the State Key Laboratory of Reproductive Medicine and Offspring Health of Nanjing Medical University. We provide our thankfulness to the Alliance of International Science Organizations (ANSO) for their support of scholarship to international students involved in this research.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| PRRSV | Porcine Respiratory and Reproductive Syndrome Virus |
| PRRSV-V | Porcine Respiratory and Reproductive Syndrome Virus-vaccine |
| SLA | Swine Leukocyte Antigen |
| HLA | Human Leukocyte Antigen |
References
- Albina, E. Epidemiology of porcine reproductive and respiratory syndrome (PRRS): an overview. Veterinary microbiology 1997, 55, 309–316. [Google Scholar] [PubMed]
- Lalonde, C.; Provost, C.; Gagnon, C.A. Whole-genome sequencing of porcine reproductive and respiratory syndrome virus from field clinical samples improves the genomic surveillance of the virus. Journal of clinical microbiology 2020, 58. [Google Scholar] [CrossRef]
- Neumann, E.J.; et al. Assessment of the economic impact of porcine reproductive and respiratory syndrome on swine production in the United States. Journal of the American Veterinary Medical Association 2005, 227, 385–392. [Google Scholar]
- Renken, C.; et al. Application of an economic calculator to determine the cost of porcine reproductive and respiratory syndrome at farm-level in 21 pig herds in Germany. Porcine health management 2021, 7, 1–12. [Google Scholar]
- Sun, Q.; et al. Emergence of a novel PRRSV-1 strain in mainland China: A recombinant strain derived from the two commercial modified live viruses Amervac and DV. Frontiers in Veterinary Science 2022, 9, 974743. [Google Scholar]
- Yu, F.; et al. Genomic analysis of porcine reproductive and respiratory syndrome virus 1 revealed extensive recombination and potential introduction events in China. Veterinary Sciences 2022, 9, 450. [Google Scholar]
- Yu, F.; et al. Phylogenetics, genomic recombination, and NSP2 polymorphic patterns of porcine reproductive and respiratory syndrome virus in China and the United States in 2014–2018. Journal of virology 2020, 94. [Google Scholar] [CrossRef]
- Wang, L.-j.; et al. Genomic analysis of a recombinant NADC30-like porcine reproductive and respiratory syndrome virus in China. Virus Genes 2018, 54, 86–97. [Google Scholar]
- Zhao, K.; et al. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. Journal of virology 2015, 89, 10712–10716. [Google Scholar]
- Ruedas-Torres, I.; et al. The jigsaw of PRRSV virulence. Veterinary Microbiology 2021, 260, 109168. [Google Scholar]
- Fang, Y.; Snijder, E.J. The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus research 2010, 154, 61–76. [Google Scholar]
- Dea, S.; et al. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: comparison of the North American and European isolates. Archives of virology 2000, 145, 659–688. [Google Scholar] [PubMed]
- Nan, Y.; et al. Improved vaccine against PRRSV: current progress and future perspective. Frontiers in microbiology 2017, 8, 1635. [Google Scholar]
- Guan, Z.; et al. Secondary Highly Pathogenic Porcine Reproductive and Respiratory Syndrome Virus (HP-PRRSV2) Infection Augments Inflammatory Responses, Clinical Outcomes, and Pathogen Load in Glaesserella-parasuis-Infected Piglets. Veterinary Sciences 2023, 10, 365. [Google Scholar]
- Zhang, H.; et al. Research Progress on the Development of Porcine Reproductive and Respiratory Syndrome Vaccines. Veterinary Sciences 2023, 10, 491. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, W. Current Status of Porcine Reproductive and Respiratory Syndrome Vaccines. Vaccines 2024, 12, 1387. [Google Scholar] [CrossRef]
- Charerntantanakul, W. Porcine reproductive and respiratory syndrome virus vaccines: Immunogenicity, efficacy and safety aspects. World journal of virology 2012, 1, 23. [Google Scholar]
- Renukaradhya, G.J.; et al. Inactivated and subunit vaccines against porcine reproductive and respiratory syndrome: current status and future direction. Vaccine 2015, 33, 3065–3072. [Google Scholar]
- Chae, C. Commercial PRRS modified-live virus vaccines. Vaccines 2021, 9, 185. [Google Scholar] [CrossRef]
- Lee, M.-A.; et al. Molecular characterization of porcine reproductive and respiratory syndrome virus in Korea from 2018 to 2022. Pathogens 2023, 12, 757. [Google Scholar] [CrossRef]
- Papatsiros, V. Impact of a killed PRRSV vaccine on sow longevity in a PRRSV infected swine herd. Journal of Applied Animal Research 2012, 40, 297–304. [Google Scholar] [CrossRef]
- Kim, H.; et al. The assessment of efficacy of porcine reproductive respiratory syndrome virus inactivated vaccine based on the viral quantity and inactivation methods. Virology journal 2011, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuwstadt, A.; et al. Proteins encoded by open reading frames 3 and 4 of the genome of Lelystad virus (Arteriviridae) are structural proteins of the virion. Journal of virology 1996, 70, 4767–4772. [Google Scholar] [CrossRef]
- Cancel-Tirado, S.M.; Evans, R.B.; Yoon, K.-J. Monoclonal antibody analysis of porcine reproductive and respiratory syndrome virus epitopes associated with antibody-dependent enhancement and neutralization of virus infection. Veterinary immunology and immunopathology 2004, 102, 249–262. [Google Scholar] [CrossRef]
- Delputte, P.; et al. Effect of virus-specific antibodies on attachment, internalization and infection of porcine reproductive and respiratory syndrome virus in primary macrophages. Veterinary immunology and immunopathology 2004, 102, 179–188. [Google Scholar] [CrossRef]
- Delputte, P.L.; et al. Porcine arterivirus attachment to the macrophage-specific receptor sialoadhesin is dependent on the sialic acid-binding activity of the N-terminal immunoglobulin domain of sialoadhesin. Journal of virology 2007, 81, 9546–9550. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; et al. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. Journal of virology 2002, 76, 4241–4250. [Google Scholar] [CrossRef]
- Plagemann, P.; Rowland, R.; Faaberg, K. The primary neutralization epitope of porcine respiratory and reproductive syndrome virus strain VR-2332 is located in the middle of the GP5 ectodomain. Archives of virology 2002, 147, 2327–2347. [Google Scholar] [CrossRef]
- Fan, B.; et al. Influence of the amino acid residues at 70 in M protein of porcine reproductive and respiratory syndrome virus on viral neutralization susceptibility to the serum antibody. Virology journal 2016, 13, 1–11. [Google Scholar] [CrossRef]
- Vanhee, M.; et al. A variable region in GP4 of European-type porcine reproductive and respiratory syndrome virus induces neutralizing antibodies against homologous but not heterologous virus strains. Viral immunology 2010, 23, 403–413. [Google Scholar] [CrossRef]
- Costers, S.; et al. GP4 of porcine reproductive and respiratory syndrome virus contains a neutralizing epitope that is susceptible to immunoselection in vitro. Archives of virology 2010, 155, 371–378. [Google Scholar] [CrossRef]
- Costers, S.; et al. GP4-specific neutralizing antibodies might be a driving force in PRRSV evolution. Virus research 2010, 154, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Wissink, E.; et al. Envelope protein requirements for the assembly of infectious virions of porcine reproductive and respiratory syndrome virus. Journal of virology 2005, 79, 12495–12506. [Google Scholar] [CrossRef] [PubMed]
- Das, P.B.; et al. The minor envelope glycoproteins GP2a and GP4 of porcine reproductive and respiratory syndrome virus interact with the receptor CD163. Journal of virology 2010, 84, 1731–1740. [Google Scholar] [CrossRef]
- Meulenberg, J.; et al. Posttranslational processing and identification of a neutralization domain of the GP4 protein encoded by ORF4 of Lelystad virus. Journal of virology 1997, 71, 6061–6067. [Google Scholar] [CrossRef]
- Vashisht, K.; et al. Identification of immunodominant T-cell epitopes present in glycoprotein 5 of the North American genotype of porcine reproductive and respiratory syndrome virus. Vaccine 2008, 26, 4747–4753. [Google Scholar] [CrossRef]
- Díaz, I.; et al. In silico prediction and ex vivo evaluation of potential T-cell epitopes in glycoproteins 4 and 5 and nucleocapsid protein of genotype-I (European) of porcine reproductive and respiratory syndrome virus. Vaccine 2009, 27, 5603–5611. [Google Scholar] [CrossRef]
- Luo, Q.; et al. Research progress on glycoprotein 5 of porcine reproductive and respiratory syndrome virus. Animals 2023, 13, 813. [Google Scholar] [CrossRef]
- Miao, Y.-Q.; et al. RETRACTED ARTICLE: N (6)-adenosine-methyltransferase-14 promotes glioma tumorigenesis by repressing argininosuccinate synthase 1 expression in an m6A-dependent manner. Bioengineered 2022, 13, 1858–1871. [Google Scholar] [CrossRef]
- Gong, H.-R.; et al. Non-neutralizing epitopes shade neutralizing epitopes against omicron in a multiple epitope-based vaccine. ACS Infectious Diseases 2022, 8, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- Devi, A.; Chaitanya, N.S. In silico designing of multi-epitope vaccine construct against human coronavirus infections. Journal of Biomolecular Structure and Dynamics 2021, 39, 6903–6917. [Google Scholar]
- Ullah, H.; et al. An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes. Biology 2024, 13, 681. [Google Scholar] [CrossRef]
- Yuan, L.; et al. In silico design of a broad-spectrum multiepitope vaccine against influenza virus. International Journal of Biological Macromolecules 2024, 254, 128071. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution 1987, 4, 406–425. [Google Scholar] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 2004, 32, 1792–1797. [Google Scholar]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics 2003, 1–22. [Google Scholar] [CrossRef]
- Edgar, R.C.; Batzoglou, S. Multiple sequence alignment. Current opinion in structural biology 2006, 16, 368–373. [Google Scholar]
- Oliver, T.; et al. Using reconfigurable hardware to accelerate multiple sequence alignment with ClustalW. Bioinformatics 2005, 21, 3431–3432. [Google Scholar]
- Jespersen, M.C.; et al. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic acids research 2017, 45, W24–W29. [Google Scholar]
- Middleton, D.; et al. New allele frequency database: http://www.allelefrequencies.net. Tissue antigens 2003, 61, 403–407. [Google Scholar] [PubMed]
- Reynisson, B.; et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic acids research 2020, 48, W449–W454. [Google Scholar]
- Calis, J.J.; et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS computational biology 2013, 9, e1003266. [Google Scholar]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC bioinformatics 2007, 8, 1–7. [Google Scholar]
- Dimitrov, I.; et al. AllerTOP v. 2—a server for in silico prediction of allergens. Journal of molecular modeling 2014, 20, 1–6. [Google Scholar]
- Gupta, S.; et al. In silico approach for predicting toxicity of peptides and proteins. PloS one 2013, 8, e73957. [Google Scholar]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biology direct 2013, 8, 1–15. [Google Scholar]
- Dhanda, S.K.; et al. Prediction of IL4 Inducing Peptides. Journal of Immunology Research 2013, 2013, 263952. [Google Scholar]
- Fadaka, A.O.; et al. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Scientific reports 2021, 11, 19707. [Google Scholar]
- Li, W.; et al. Peptide vaccine: progress and challenges. Vaccines 2014, 2, 515–536. [Google Scholar] [CrossRef]
- Jalal, K.; et al. Pan-genome reverse vaccinology approach for the design of multi-epitope vaccine construct against Escherichia albertii. International Journal of Molecular Sciences 2021, 22, 12814. [Google Scholar] [PubMed]
- Dhanushkumar, T.; et al. Structural immunoinformatics approach for rational design of a multi-epitope vaccine against triple negative breast cancer. International Journal of Biological Macromolecules 2023, 243, 125209. [Google Scholar]
- Ghaffari-Nazari, H.; et al. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T help in BALB/c mice. PloS one 2015, 10, e0142563. [Google Scholar]
- Gasteiger, E.; et al. Protein Analysis Tools on the ExPASy Server 571 571 From: The Proteomics Protocols Handbook Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook 2019, 571–607. [Google Scholar]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [PubMed]
- Kelley, L.A.; et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nature protocols 2015, 10, 845–858. [Google Scholar] [PubMed]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic acids research 2013, 41, W384–W388. [Google Scholar]
- Wiederstein, M.; Sippl, M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic acids research 2007, 35 (Suppl. 2), W407–W410. [Google Scholar] [CrossRef]
- Laskowski, R.A.; et al. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of applied crystallography 1993, 26, 283–291. [Google Scholar]
- Liu, D.; Chen, Y. Epitope screening and vaccine molecule design of PRRSV GP3 and GP5 protein based on immunoinformatics. Journal of Cellular and Molecular Medicine 2024, 28, e18103. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.A.; Sarkar, B.; Islam, S.S. Exploiting the reverse vaccinology approach to design novel subunit vaccines against Ebola virus. Immunobiology 2020, 225, 151949. [Google Scholar] [CrossRef]
- Yan, Y.; et al. The HDOCK server for integrated protein–protein docking. Nature protocols 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; et al. PDBsum: Structural summaries of PDB entries. Protein science 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Structure Function and Bioinformatics 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Case, D.; et al. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Maier, J.A.; et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of chemical theory and computation 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; et al. Development and testing of a general amber force field. Journal of computational chemistry 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of computational chemistry 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. Journal of chemical theory and computation 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40, 82–92. [Google Scholar]
- Rapin, N.; et al. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PloS one 2010, 5, e9862. [Google Scholar] [CrossRef] [PubMed]
- Grote, A.; et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research 2005, 33 (Suppl. 2), W526–W531. [Google Scholar] [CrossRef]
- Ullah, H.; et al. An In Silico Design of a Vaccine against All Serotypes of the Dengue Virus Based on Virtual Screening of B-Cell and T-Cell Epitopes. Biology 2024, 13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
An overview of the reverse vaccinology process used to design the Porcine reproductive and respiratory syndrome virus vaccine. Data collection, epitope prediction, vaccine candidate construction, and in silico validation are highlighted.
Figure 1.
An overview of the reverse vaccinology process used to design the Porcine reproductive and respiratory syndrome virus vaccine. Data collection, epitope prediction, vaccine candidate construction, and in silico validation are highlighted.
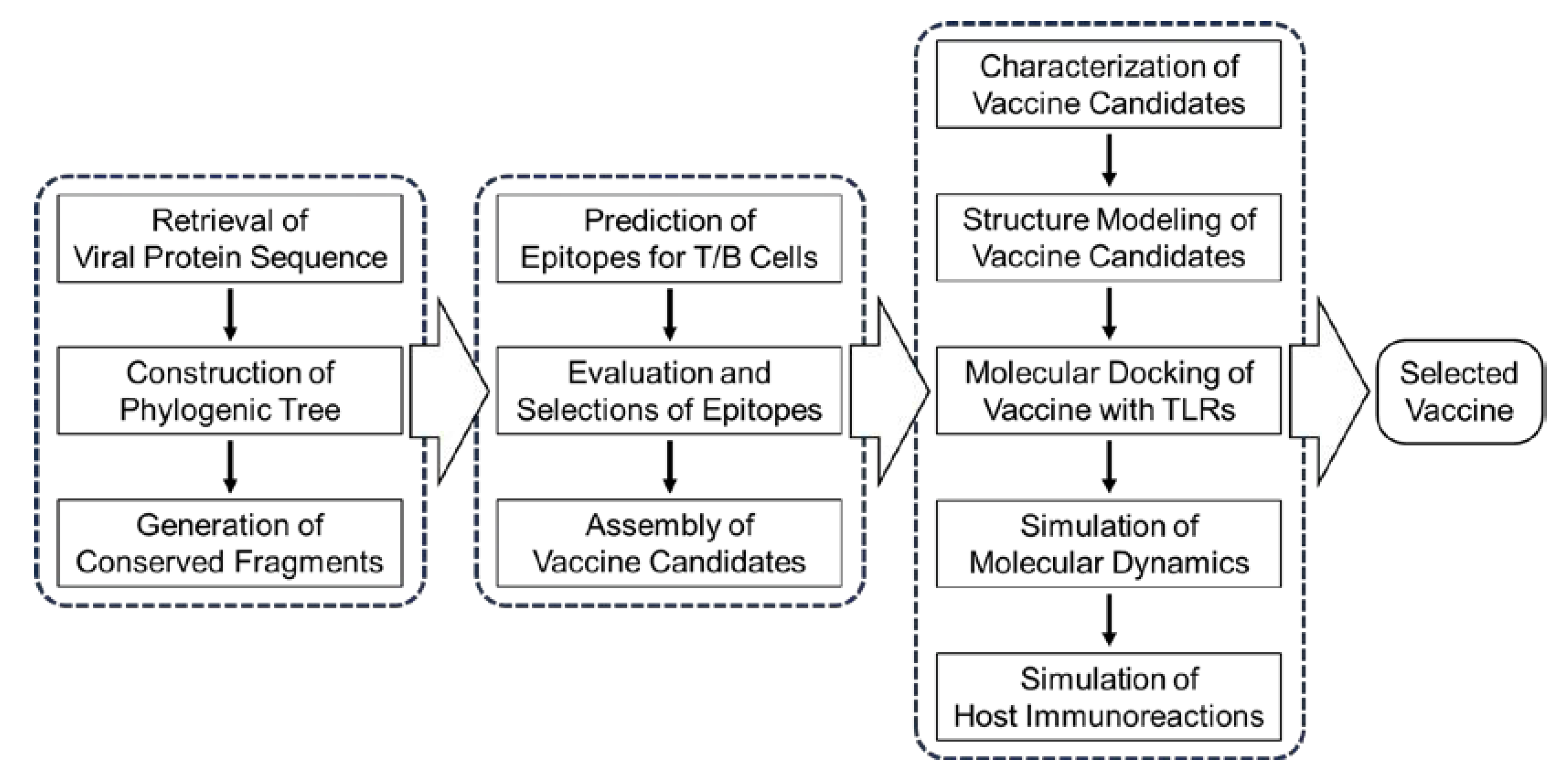
Figure 2.
Organization of multi-epitope vaccine peptides. (A)graphic representation of the vaccine design approach that includes the core antigen and summary of the approach taken to create the multiepitope vaccine peptide. This design includes specific B cells, CTLs, and HTLs epitopes that are bordered by Adjuvant and connected by peptide linkers. (B-D) Vaccine candidates of PRRSV-V-1, PRRSV-V-2, PRRSV-V-3 (B) PRRSV-V-1, Core antigen conjugated with S50 L7/12 ribosomal protein at the N-terminal, (C) PRRSV-V-2, at the N-terminal β-defensin is attached to the core antigen (D) HBHA adjuvant is conjugated with core antigen at the N terminal.
Figure 2.
Organization of multi-epitope vaccine peptides. (A)graphic representation of the vaccine design approach that includes the core antigen and summary of the approach taken to create the multiepitope vaccine peptide. This design includes specific B cells, CTLs, and HTLs epitopes that are bordered by Adjuvant and connected by peptide linkers. (B-D) Vaccine candidates of PRRSV-V-1, PRRSV-V-2, PRRSV-V-3 (B) PRRSV-V-1, Core antigen conjugated with S50 L7/12 ribosomal protein at the N-terminal, (C) PRRSV-V-2, at the N-terminal β-defensin is attached to the core antigen (D) HBHA adjuvant is conjugated with core antigen at the N terminal.
Figure 3.
Modeling and validation of vaccine candidates’ molecular structure. (A) Composition of secondary structure for PRRSV-V-1~ PRRSV-V-3, showing the percentage distribution of coils, helices, and β-strands in each protein. (B) Confidence level for the tertiary structure models of PRRSV-V-1~ PRRSV-V-3. (C) Coverage level of tertiary structure models PRRSV-V-1~ PRRSV-V-3. (D) According to Ramachandran plots, the distribution of residues in favored, allowed, and disallowed regions of PRRSV-V-1~ PRRSV-V-3. (E) Z-score of PRRSV-V-1~3, according to their Z-score plots.
Figure 3.
Modeling and validation of vaccine candidates’ molecular structure. (A) Composition of secondary structure for PRRSV-V-1~ PRRSV-V-3, showing the percentage distribution of coils, helices, and β-strands in each protein. (B) Confidence level for the tertiary structure models of PRRSV-V-1~ PRRSV-V-3. (C) Coverage level of tertiary structure models PRRSV-V-1~ PRRSV-V-3. (D) According to Ramachandran plots, the distribution of residues in favored, allowed, and disallowed regions of PRRSV-V-1~ PRRSV-V-3. (E) Z-score of PRRSV-V-1~3, according to their Z-score plots.
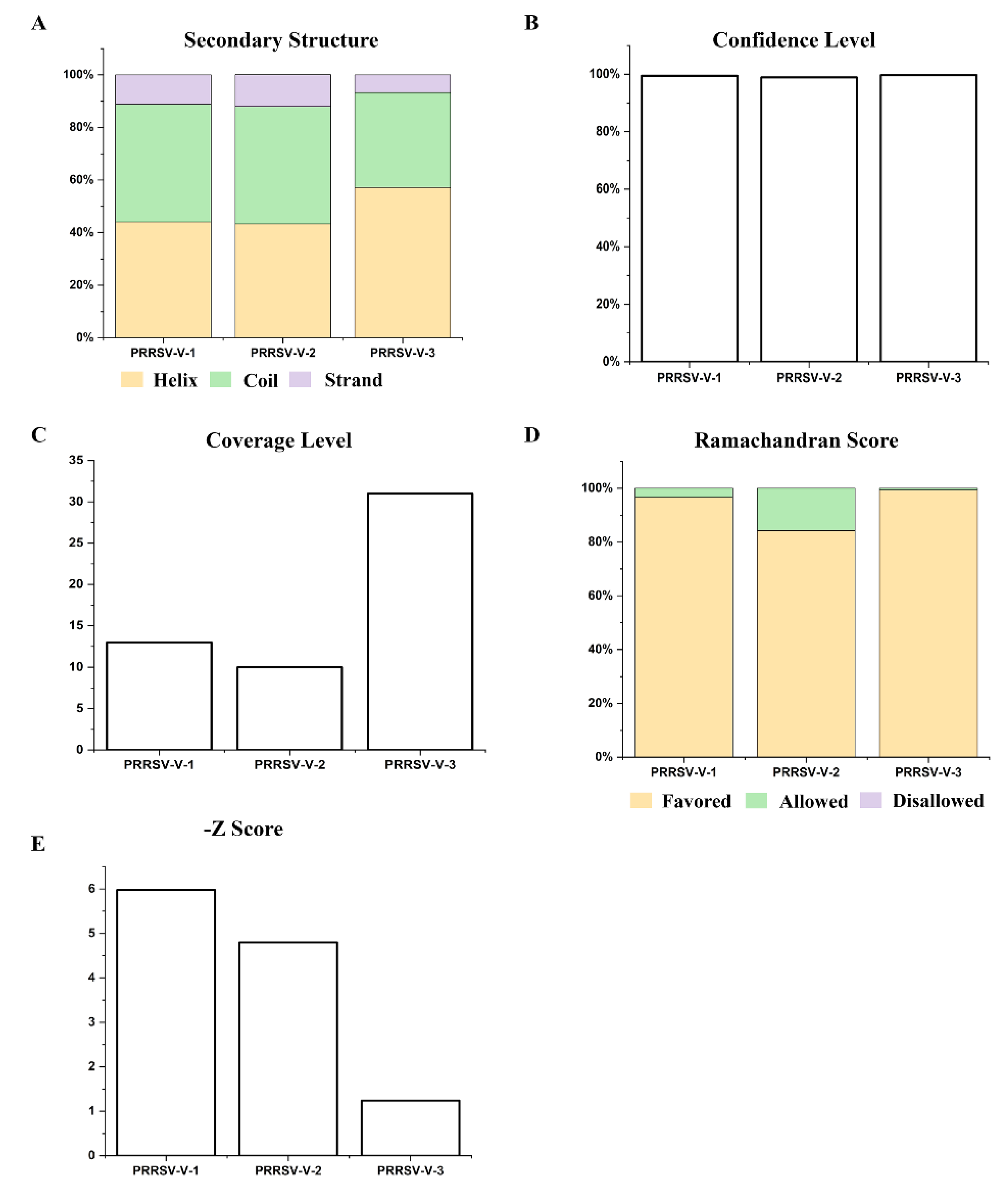
Figure 4.
Molecular Docking, interaction analysis and Molecular dynamic simulations of PRRSV-V-8 with TLR3/TLR8. (A) comprehensive statistics summary of the interface properties between PRRSV-V-1~ PRRSV-V-3with TLR3/TLR8 (B)Root means square deviation (RMSD): shows the RMSD for the complexes of PRRSV-V-2 with TLR3 and TLR8. (C)Radius of Gyration (Rg): Rg the compactness of PRRSV-V-2 with TLR3 and TLR8. (D)Hydrogen Bonds: the count of intra-molecule hydrogen bonds formed of PRRSV-V-2 with TLR3 and TLR8.graphical representations of dynamics simulation of PRRSV-V-2 with TLR3 are indicated in Red, whereas those dynamics simulation of PRRSV-V-2 with TLR8 are shown in Black.
Figure 4.
Molecular Docking, interaction analysis and Molecular dynamic simulations of PRRSV-V-8 with TLR3/TLR8. (A) comprehensive statistics summary of the interface properties between PRRSV-V-1~ PRRSV-V-3with TLR3/TLR8 (B)Root means square deviation (RMSD): shows the RMSD for the complexes of PRRSV-V-2 with TLR3 and TLR8. (C)Radius of Gyration (Rg): Rg the compactness of PRRSV-V-2 with TLR3 and TLR8. (D)Hydrogen Bonds: the count of intra-molecule hydrogen bonds formed of PRRSV-V-2 with TLR3 and TLR8.graphical representations of dynamics simulation of PRRSV-V-2 with TLR3 are indicated in Red, whereas those dynamics simulation of PRRSV-V-2 with TLR8 are shown in Black.
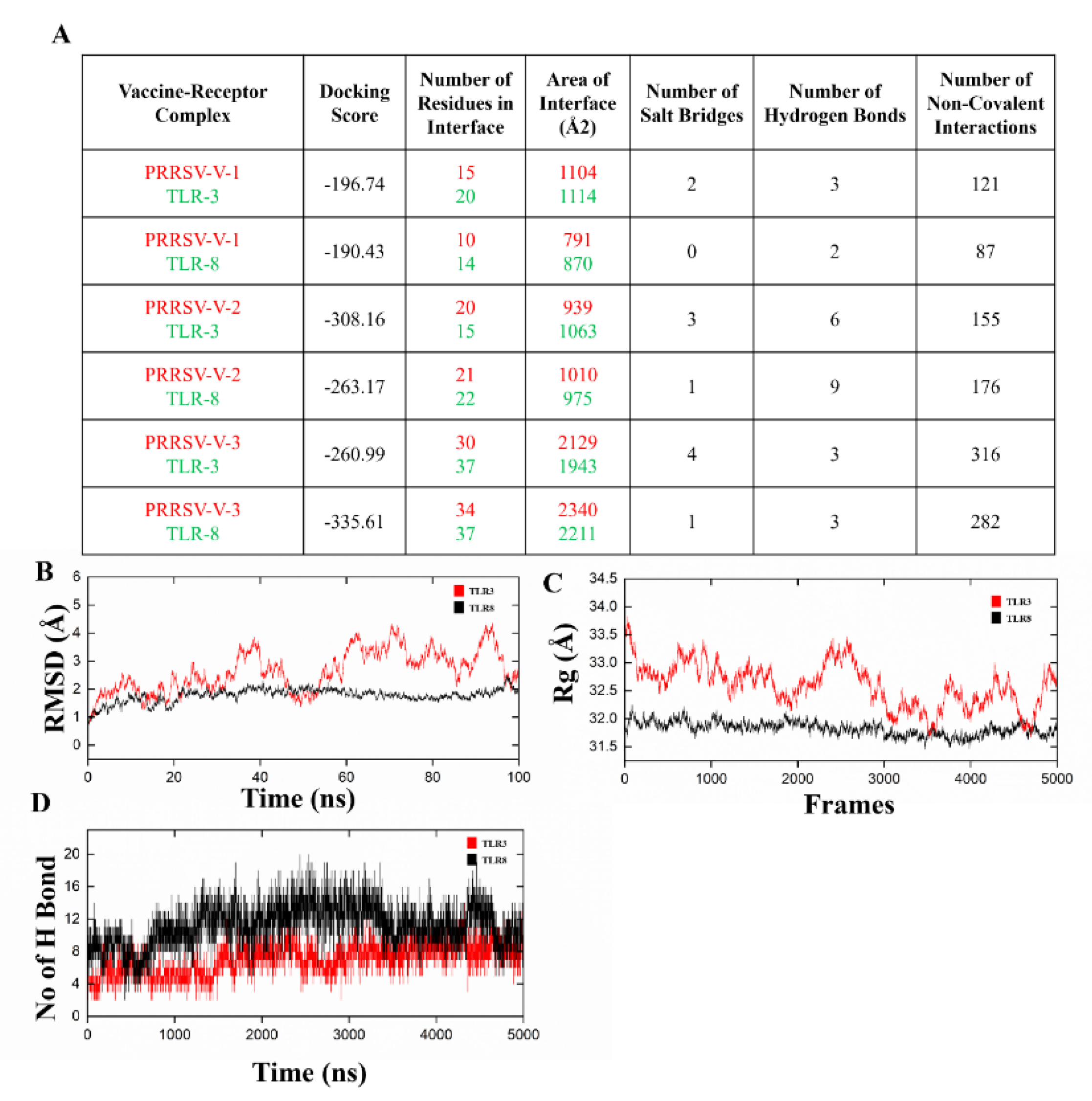
Figure 5.
Simulation of innate immune response spots PRRSV-V-2 vaccination. (A) kinetics of important cytokine expression levels after three PRRSV-V-2 dosages. There were included TGF- β, TNF-alpha, IL-2, IL4, IL6, IL-10, IL12, IL18, IL-23, IFN-gamma, and IFN- β. The Blue arrow denoted the IFN-gamma peak level. (B) Dendritic cell (DC) responses following three PRRSV-V-2 doses. the DC population as a whole and its subpopulation are shown. internalized, presenting-1, presenting-2, active, and resting DCs were among the subpopulations. (C) Macrophages (MA) reactions to three doses of PRRSV-V-2 vaccination. The macrophage (MA) population as a whole and its subpopulations are depicted. internalized, presenting-1, presenting-2, active, and resting MA were among the subgroups. (D) Natural Killer (NK) cell dynamics after vaccination.
Figure 5.
Simulation of innate immune response spots PRRSV-V-2 vaccination. (A) kinetics of important cytokine expression levels after three PRRSV-V-2 dosages. There were included TGF- β, TNF-alpha, IL-2, IL4, IL6, IL-10, IL12, IL18, IL-23, IFN-gamma, and IFN- β. The Blue arrow denoted the IFN-gamma peak level. (B) Dendritic cell (DC) responses following three PRRSV-V-2 doses. the DC population as a whole and its subpopulation are shown. internalized, presenting-1, presenting-2, active, and resting DCs were among the subpopulations. (C) Macrophages (MA) reactions to three doses of PRRSV-V-2 vaccination. The macrophage (MA) population as a whole and its subpopulations are depicted. internalized, presenting-1, presenting-2, active, and resting MA were among the subgroups. (D) Natural Killer (NK) cell dynamics after vaccination.
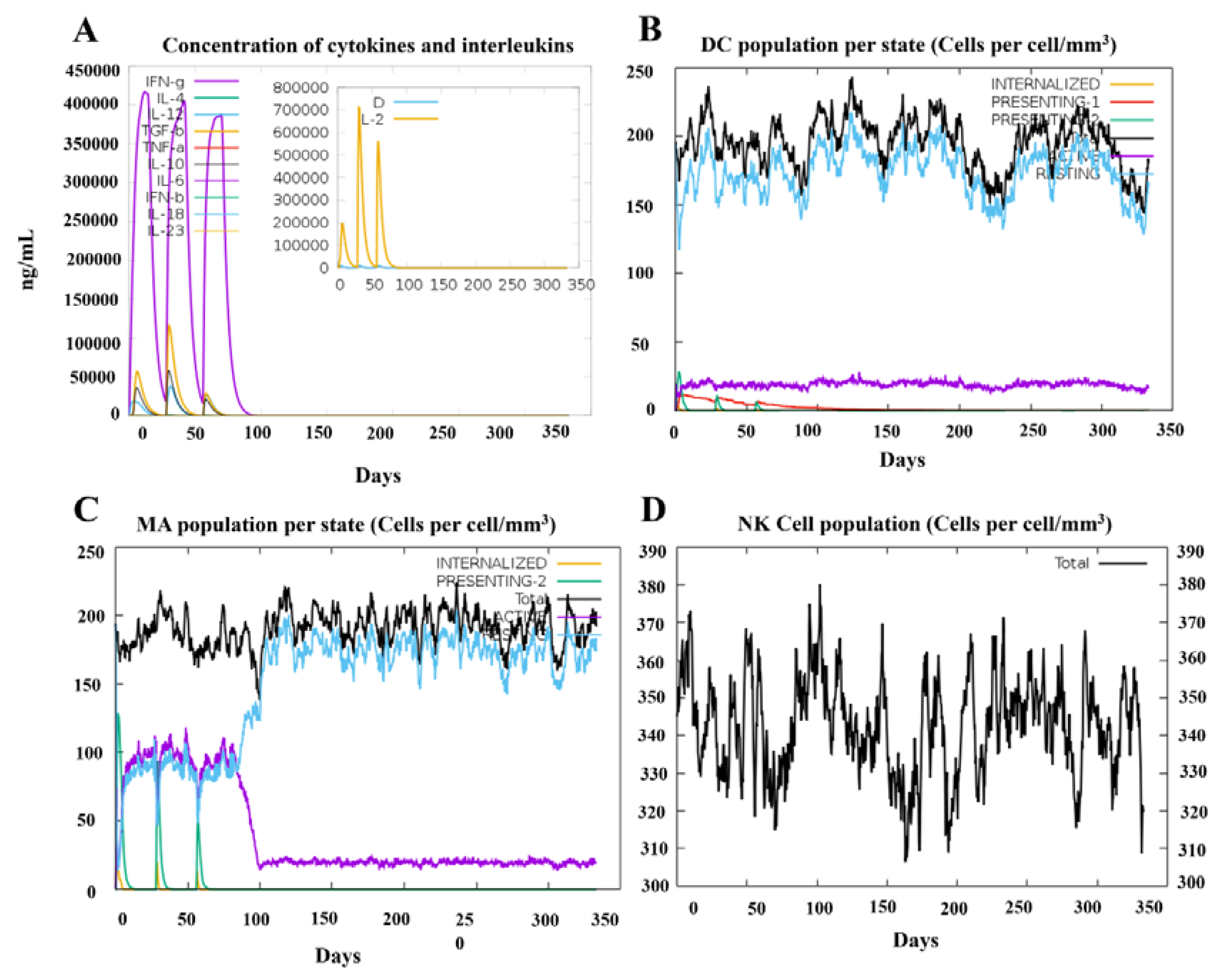

Table 1.
Prediction of B cell epitopes.
| Genotype | protein | Start -end |
epitopes | length | Antigenicity Score |
Allergenicity /Toxicity |
Intra/Inter Conservancy |
|---|---|---|---|---|---|---|---|
| PRRSV1 | ORF2A | 57-66 | APRYSVRALP | 10 | 0.8784 | Non/Non | 75.00% /20.00% |
| PRRSV2 | GP3 | 38-45 | LVRGNFSF | 8 | 1.7037 | Non/Non | 100.00% /33.33% |
| PRRSV2 | GP4 | 44-57 | QHTQQHHLVIDHIR | 14 | 0.5457 | Non/Non | 100.00% /33.33% |
| PRRSV1 | GP5 | 49-61 | LNGTDWLDKRFDW | 13 | 1.7333 | Non/Non | 75.00% /20.00% |
| PRRSV2 | M | 131-144 | RKPGLTSVNGTLV | 13 | 0.8659 | Non/Non | 100.00% /33.33% |
| PRRSV1 | N | 6-13 | HFPLAAED | 8 | 0.9417 | Non/Non | 66.00% /22.22% |
Table 2.
Prediction of CTL epitopes. Shortlisted CTL(MHC-I) epitopes along with their strain, and predicted immunogenicity, Antigenicity, Toxicity, and conserve analysis.
Table 2.
Prediction of CTL epitopes. Shortlisted CTL(MHC-I) epitopes along with their strain, and predicted immunogenicity, Antigenicity, Toxicity, and conserve analysis.
| Genotype | protein | SLA Allele | Epitope | Antigenicity /Immunogenicity |
Allergenicity /Toxicity |
Intra/Inter Conservancy |
|---|---|---|---|---|---|---|
| PRRSV1 | ORF1a | 1*1101 | AALTGRTL | 1.2071 /0.14994 |
Non/Non | 100.00% /25.00% |
| PRRSV1 | ORF1a | 3*0501 | HQKPIAYANL | 1.1782 /0.1123 |
Non/Non | 100.00% /20.00% |
| PRRSV2 | ORF2b | 3*0701 | TRARHAIF | 1.0977 /0.31383 |
Non/Non | 95.24% /25.00% |
| PRRSV2 | GP2a | 3*0401 | SVRALPFTL | 1.5046 /0.15641 |
Non/Non | 75.00% /22.22% |
| PRRSV1 | GP2b | 2*0101 | IFLAILFGF | 0.7123 /0.27353 |
Non/Non | 75.00% /22.22% |
| PRRSV2 | GP3 | 2*0101 | YAWLAFLSF | 1.1215 /0.10302 |
Non/Non | 100.00% /22.22% |
| PRRSV2 | GP4 | 1*0702 | SACVNFTDY | 1.6035 /0.17366 |
Non/Non | 66.67% /22.22% |
| PRRSV2 | GP5 | 3*0101 | TRYTNFLL | 1.2753 /0.1324 |
Non/Non | 66.67% /25.00% |
| PRRSV1 | M | 2*1201 | FSITYTPII | 1.4228 /0.1836 |
Non/Non | 66.67% /22.22% |
| PRRSV1 | N | 2*1101 | FPLATEDDVRHHF | 0.4645 /0.34939 |
Non/Non | 50.00% /38.46% |
Table 3.
Prediction of HTL epitopes: Predicted antigenicity, Interferon induction, toxicity, and conservancy analysis of shortlisted HTL(MHC-II) epitopes.
Table 3.
Prediction of HTL epitopes: Predicted antigenicity, Interferon induction, toxicity, and conservancy analysis of shortlisted HTL(MHC-II) epitopes.
| Genotype | Protein | HLA- Allele | Epitope | Antigenicity | Allergenicity/toxicity | IL-4/IFN- γ induction | Intra/Inter Conservancy |
|---|---|---|---|---|---|---|---|
| PRRSV1 | ORF1 | DPA1*02:01 DPB1*05:01 |
GVAPAVRIAERYRGR | 0.9596 | Non/Non | Inducer /Inducer |
100.00% /13.33% |
| PRRSV1 | ORF | DRB4*01:04 | KPIAYANLDEKKISA | 0.9123 | Non/Non | Inducer /Inducer |
100.00% /13.33% |
| PRRSV2 | ORF1b | DRB1*12:04 | CLGDFKQLHPVGFDS | 1.2326 | Non/Non | Inducer /Inducer |
95.24% /20.00% |
| PRRSV2 | GP2a | DPA1*02:01 DPB1*04:01 |
LSFASDWFAPRYSVR | 0.5461 | Non/Non | Inducer /Inducer |
75.00% /20.00% |
| PRRSV2 | GP2b | DRB1*04:04 | VFCIRLVCSAIHRSR | 1.3347 | Non/Non | Inducer /Inducer |
100.00% /20.00% |
| PRRSV2 | GP3 | DPA1*02:01 DPB1*04:01 |
NWFHLEWLRPFFSSW | 0.4583 | Non/Non | Inducer /Inducer |
75.00% /20.00% |
| PRRSV1 | GP4 | DRB5*01:03 | ACVNFTDYVAHVTQH | 1.0257 | Non/Non | Inducer /Inducer |
75.00% /13.33% |
| PRRSV2 | GP5 | DPA1*01:03 DPB1*01:01 |
WRYSCTRYTNFLLDT | 0.5114 | Non/Non | Inducer /Inducer |
100.00% /13.33% |
| PRRSV1 | M | DPA1*02:01 DPB1*02:01 |
LAFSITYTPIIYALK | 1.4017 | Non/Non | Inducer /Inducer |
100.00% /13.33% |
| PRRSV2 | N | DQA1*01:01 DQB1*02:01 |
PHFPLATEDDVRHHF | 0.4807 | Non/Non | Inducer /Inducer |
66.67% /26.67% |
Table 4.
Physiochemical properties of the predicted vaccine candidate.
| Parameters | PRRSV-V-1 vaccine | PRRSV-V-2 vaccine | PRRSV-V-3 vaccine |
|---|---|---|---|
| No. of amino acids | 531 | 448 | 551 |
| Molecular weight | 57526.93 | 49446.89 | 60733.93 |
| Instability Index | 22.04 | 25.72 | 28.66 |
| Aliphatic index | 81.02 | 80.91 | 77.04 |
| Half-life | 30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) |
30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) |
30 h (mammalian reticulocytes, in vitro). >20 h (yeast, in vivo). >10 h (Escherichia coli, in vivo) |
| Solubility | 0.979905 | 0.975094 | 0.986037 |
| GRAVY | -0.054 | -0.191 | -0.249 |
| Theoretical pI | 9.12 | 9.82 | 9.17 |
| Antigenicity | 0.61 | 0.68 | 0.63 |
| Allergenicity | Non-Allergenic | Non-Allergenic | Non-Allergenic |
| Toxicity | Non-Toxic | Non-Toxic | Non-Toxic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



