
Submitted:
03 March 2025
Posted:
04 March 2025
You are already at the latest version
Abstract
Bacterial pathogens have evolved diverse strategies to infect hosts, evade immune responses, and establish successful infections. While the role of transcription factors in bacterial virulence is well-documented, emerging evidence highlights the significant contribution of small regulatory RNAs (sRNAs) in bacterial pathogenesis. These sRNAs function as post-transcriptional regulators that fine-tune gene expression, enabling bacteria to adapt rapidly to challenging environments. This review explores the multifaceted roles of bacterial sRNAs in host-pathogen interactions. Firstly, it examines how sRNAs regulate virulence by modulating the expression of key virulence factors, including fimbriae, toxins, and secretion systems followed by discussing the role of sRNAs in bacterial stress response mechanisms that counteract host immune defenses, such as oxidative and envelope stress resistance pathways. Additionally, this review investigates the involvement of sRNAs in antibiotic resistance by regulating efflux pumps, biofilm formation, and target modifications, which contribute to multidrug resistance phenotypes. Lastly, this review highlights how sRNAs contribute to intra- and inter-species communication through quorum sensing, thereby coordinating bacterial behavior in response to environmental cues. Understanding these regulatory networks governed by small RNAs (sRNAs) is essential for the development of innovative antimicrobial strategies. This review highlights the growing significance of sRNAs in bacterial pathogenicity and explores their potential as therapeutic targets for the treatment of bacterial infections.
Keywords:
Small RNA
; bacterial pathogenesis
; stress response
; antibiotic resistance
; quorum sensing
; host-pathogen interactions
1. Introduction
The human body harbors a complex and diverse microbiota that colonizes nearly all organs, including the gastrointestinal tract, skin, and respiratory system. Bacterial cells are estimated to outnumber human cells by approximately tenfold. While many of these microorganisms contribute to host health through symbiotic relationships, humans are also frequently exposed to pathogenic bacteria capable of establishing infections. The human host employs multiple strategies to curb bacterial infections, including physical barriers, immune responses, and antimicrobial molecules [1]. For example, the skin and mucosal surfaces serve as the first line of defense, preventing bacterial entry through tight junctions and mucus secretion, which entraps pathogens such as Pseudomonas aeruginosa [2]. Innate immune responses further combat infections through the production of antimicrobial peptides (AMPs) like defensins and cathelicidins, which disrupt bacterial membranes, effectively targeting pathogens such as Staphylococcus aureus [3, 4], Mycobacterium tuberculosis and Human Immunodeficiency Virus (HIV) [5]. The complement system enhances bacterial clearance by opsonization, and membrane attack complex formation, particularly against Neisseria meningitidis [6]. Additionally, host cells limit bacterial growth by sequestering essential nutrients, a strategy known as nutritional immunity; for instance, lactoferrin and calprotectin bind iron and zinc to restrict the proliferation of uropathogenic strains of Escherichia coli and Klebsiella pneumoniae in the urinary tract [7-9]. Adaptive immunity further strengthens bacterial control through antibody production and T-cell-mediated responses, which are critical for long-term protection against Mycobacterium tuberculosis [10]. These multilayered defense mechanisms collectively work to prevent bacterial infections and maintain host homeostasis. To circumvent these host defenses and ensure their survival inside the host and establish an infection, the bacterial pathogens have evolved sophisticated virulence factors and immune evasion strategies [11-13]. These include spatiotemporal expression of virulence factors as well as stress responsive pathways that equips the pathogen with the necessary adaptations needed to colonize and persist within the host. In addition, bacterial pathogens often communicate with each other through chemical messengers which are collectively called quorum sensing [14-17]. Bacterial quorum sensing regulates group behaviors such as biofilm formation, virulence factor expression, and antibiotic resistance. By sensing and responding to host-derived signals, bacteria can coordinate adaptive responses that enhance colonization, persistence, and immune evasion. Finally, the bacteria have evolved a series of strategies to curb the effect of antibiotics including modification of lipid bilayer, expression of efflux pumps and inhibition of antibiotic uptake systems [18-20]. Together, the bacteria have developed a sophisticated system to evade host immunity, and studying the molecular mechanisms behind this process could lead to new therapeutic advancements. Although, the role of various proteins in virulence of these pathogens have been well characterized, the importance of bacterial small regulatory RNA was only recognized in the last decade or so.
Bacterial small regulatory RNAs (sRNAs) are a diverse class of molecules, typically ranging from 40 to 500 nucleotides, that play essential roles in post-transcriptional gene regulation by interacting with target mRNAs or proteins. Initially categorized as small non-coding RNAs, some sRNAs, such SgrS from enteric species, SR1 from Bacillus subtilis, , Pel RNA from Streptococcus pyogenes, RNAIII and Psm-mec from Staphylococcus aureus have been found to encode small peptides [21], leading to their classification as small regulatory RNAs. sRNAs are involved in key cellular processes, including regulation of bacteria transport through modulation of outer membrane protein expression, iron homeostasis, quorum sensing, and bacterial virulence [22-26]. Advancements in bioinformatics and experimental methodologies have facilitated the discovery of over 900 bacterial sRNAs, primarily transcribed from intergenic regions [27]. These sRNAs are classified as either cis-encoded or trans-encoded based on their genomic arrangement relative to target genes. Cis-encoded sRNAs are transcribed from the same genomic locus as their target mRNA and typically exhibit perfect complementarity. In contrast, trans-encoded sRNAs are transcribed from separate loci and regulate multiple targets with partial complementarity, often requiring RNA chaperones like Hfq [28]. Computational approaches, including comparative genomics and machine learning-based predictions, have significantly improved sRNA identification and target prediction, although experimental validation remains essential due to challenges posed by imperfect base pairing in trans-encoded sRNAs. The continued discovery and functional characterization of sRNAs are critical to understanding their roles in bacterial stress response and pathogenesis [29].
Bacteria encounter numerous stressors within the host environment, including nutrient deprivation, temperature fluctuations, oxidative stress, osmotic pressure changes, and exposure to antimicrobial compounds [30, 31]. To mitigate the harmful effects of the aforementioned host response, bacteria have developed intricate stress response pathways regulated through transcriptional control by sigma factors and two-component systems. [32-34]. The discovery of sRNAs has unveiled an additional layer of post-transcriptional regulation, enabling bacteria to fine-tune gene expression with high precision and efficiency. These small RNAs regulate the expression of their target gene by altering mRNA stability, translation, and processing, allowing rapid adaptation to environmental changes. By coordinating cellular processes such as metabolic reprogramming [35, 36], detoxification [37], DNA repair [38], and biofilm formation [39, 40], sRNAs serve as pivotal regulators of bacterial stress responses. Beyond stress adaptation, sRNAs also play a central role in bacterial virulence, governing the expression of key pathogenicity determinants, including toxins [41], adhesins, invasins [42], and immune evasion factors [43]. Furthermore, sRNAs have been shown to play a key role in antimicrobial resistance, one of the pressing global concerns.
The rise of antimicrobial resistance (AMR) represents a significant global health threat, with resistant infections leading to increased morbidity, mortality, and healthcare costs [44]. The World Health Organization (WHO) has identified 24 priority pathogens exhibiting significant resistance, including multidrug-resistant Mycobacterium tuberculosis and Gram-negative bacteria resistant to last-line antibiotics [45]. Projections indicate that AMR could result in approximately 39 million deaths between 2025 and 2050, underscoring the urgency of addressing this crisis.
While mechanisms such as horizontal gene transfer and mutations in antibiotic target sites have been extensively studied [46, 47], recent research has identified sRNAs as important contributors to antimicrobial resistance [48, 49]. These regulatory RNAs influence antibiotic resistance through multiple pathways including modification of cell membrane components, repression of antibiotic uptake systems, activation of efflux pumps, and generation of biofilms. In addition, bacteria can also achieve antimicrobial resistance through an alternative mechanism—quorum sensing. This process enables bacteria to respond to specific chemical messengers by fine-tuning the expression of genes involved in biofilm formation and antimicrobial resistance, allowing them to adapt to changing environmental conditions. This dual functionality highlights the versatility of small RNAs in shaping bacterial survival and community dynamics.
Bacteria exist within complex microbial communities, where interspecies communication is essential for survival with the host. Quorum sensing is a bacterial communication process that enables populations to coordinate gene expression based on cell density. This signaling mechanism relies on the production, release, and detection of small signaling molecules called autoinducers. As bacterial populations grow, the concentration of these molecules increases, allowing bacteria to sense their community size and regulate various behaviors collectively. Quorum sensing controls a wide range of functions, including biofilm formation, virulence factor production, antibiotic resistance, and bioluminescence. By synchronizing gene expression, quorum sensing enables bacteria to behave as a unified group, enhancing their survival and adaptability in diverse environments. [15, 16]. Emerging evidence suggests that sRNAs plays a key role in regulation of quorum sensing [50, 51]. This sRNA-mediated communication extends beyond intra-species interactions, as these regulatory molecules can be transferred between bacterial species and even across domains, impacting host-microbe interactions [52, 53]. Understanding the mechanisms by which sRNAs mediate interspecies communication will provide deeper insights into microbial ecology and host-associated microbial dynamics, potentially informing strategies to manipulate microbial communities for therapeutic, agricultural, and industrial applications. This review will focus on role of bacterial sRNAs in host-pathogen interaction by governing essential processes such as stress response, virulence regulation, antibiotic resistance, and interspecies communication as outlined in the following sections.
2. sRNA as the Orchestrators of Bacterial Pathogenesis
Pathogenic bacteria acquire foreign genes through horizontal gene transfer that can be integrated into the bacterial genome (pathogenicity islands) or exists as plasmids [54]. These horizontally acquired genetic elements harbor genes involved in bacterial pathogenicity, immune evasion, and persistence. These genetic elements are also a rich hub for small RNAs [55]. sRNAs play a crucial role in bacterial pathogenesis by precisely regulating the spatiotemporal expression of bacterial genes, ensuring adaptation to dynamic host environments, as detailed below.
Attachment to the host is the first step in bacterial colonization, allowing pathogens to anchor to host surfaces and establish infection niches. Bacterial fimbriae, pili, adhesins etc. mediates these interactions, with sRNAs playing a crucial role in their regulation. For example, FimR2 sRNA controls the expression of fimbrial operons in Escherichia coli [56]. Similarly, small RNAs MicC and InvR regulates outer membrane protein OmpD in Salmonella Typhimurium and Salmonella entertidis respectively that enables bacterial adherence to macrophages [57]. This regulation ensures optimal colonization while preventing premature immune recognition. Upon adherence, bacteria deploy invasion mechanisms, often involving secretion systems that inject effector proteins into host cells. Salmonella enterica utilizes Type III Secretion Systems (T3SS) located at Salmonella pathogenicity island I (SP1) enabling epithelial cell invasion and intracellular survival [58]. sRNAs modulate the expression of these virulence genes located at SP1 in response to host-derived signals, ensuring their activation only when needed [59, 60]. Following invasion, bacterial pathogens must evade immune responses and establish a replicative niche. For example, Mycobacterium tuberculosis employs sRNAs to regulate dormancy and macrophage survival, facilitating long-term persistence [61]. In Salmonella, sRNAs govern intracellular survival within macrophages by fine-tuning stress response pathways and modifying virulence gene expression [62]. Additionally, sRNAs contribute to iron uptake, quorum sensing, and biofilm formation, all of which are essential for bacterial persistence and dissemination as described in the details in this section.
2.1. Adherence to the Host
Uropathogenic Escherichia coli (UPEC) encode at least nine types of fimbriae including Type 1, P, S-fimbriae, and afimbrial adhesin I, which are encoded by the fim, pap, sfa, and afa operons, respectively [63, 64] (Table S1). Among these, Type 1 fimbriae is the most prevalent, with more than 90% of both commensal and pathogenic E. coli strains harboring this adhesin [65]. In contrast, P-fimbriae, S-fimbriae, and afimbrial adhesin I are present in approximately 40–60%, 30–60%, and 0–12.5% of E. coli isolates, respectively [66]. Type 1 fimbriae play a crucial role in UPEC pathogenesis, as they facilitate adherence to and invasion of bladder epithelial cells, allowing intracellular replication and the formation of intracellular bacterial communities (IBCs) [12,13,14]. These fimbriae, encoded by the fimAICDFGH operon, plays a crucial role in bacterial colonization, particularly in murine models of urinary tract infection (UTI). The expression of fim is regulated by multiple transcription factors, including Histone like Nucleoid Structuring protein, H-NS, Factor for inversion stimulation, Fis, and Leucine responsive regulatory protein, Lrp. Additionally, a 314-base pair inverted repeat element located upstream of fimA functions as a phase-variable switch (fimS), existing in either an ON or OFF state, controls the expression of the fimbrial operon. The inversion of the fimS switch is mediated by site-specific recombinases such as FimB and FimE, as well as IpuA, IpuB, and IpbA [67]. P fimbriae enhance the virulence of UPEC by facilitating adhesion to uroepithelial cells in both in vitro and in vivo settings. Uropathogenic Escherichia coli (UPEC) is the primary causative agent of urinary tract infections (UTIs). It enters the host through the urethra, ascends to the urinary bladder causing cystitis [68], and if left untreated, may progress to the kidneys, leading to pyelonephritis [69]. UPEC employs diverse virulence factors, including fimbrial adhesins, to colonize host tissues and evade immune defenses. In various Gram-negative bacteria, the chaperone–usher pathway (CUP) functions as a molecular machine that facilitates the assembly of fimbriae on the bacterial outer membrane and the adhesins located at the fimbrial tips play a crucial role in bacterial attachment to host epithelial receptors. Uropathogenic Escherichia coli (UPEC) strains encode multiple CUP operons. Several CUP-encoded fimbriae, including Auf, Dr, F1C, S, type 9, type 3, type 1, and P fimbriae, have been identified in UPEC pathotypes. However, among those broadly conserved in UPEC isolates, only type 1 and P fimbriae have been strongly associated with urinary tract infection (UTI) pathogenesis [70]. Among these, P-fimbriae play a critical role in kidney colonization by binding to globoseries glycosphingolipids on uroepithelial cells [71]. Unlike type 1 fimbriae, which mediate mannose-sensitive adhesion, P-fimbriae facilitate mannose-resistant adherence, promoting persistent infection, resistance to urinary flow, and immune evasion [63]. Recent study has elucidated the role of small RNA PapR in regulation of P-fimbriae.
2.1.1. PapR
A large-scale phylogenomic analysis of 907 E. coli isolates, including 722 UPEC strains, identified the papGII locus—encoding P fimbriae—as a key genetic determinant distinguishing invasive UPEC associated with severe UTIs, such as pyelonephritis and urinary-source bacteremia, from non-invasive UPEC strains linked to asymptomatic bacteriuria or cystitis [72]. Notably, the number of pap operon copies (papGFEKJDCHABI) varies among UPEC strains, with UTI89 harboring a single copy and CFT073 possessing two copies. Beyond their role in adhesion, P-fimbriae contribute to biofilm formation, enhancing bacterial survival by providing protection against host immune responses and antimicrobial agents [73]. The phase variation of P-fimbriae is tightly regulated through an antagonistic interaction between PapI and the small RNA PapR (Figure 1). PapR, a 180nt long sRNA was identified as a small RNA that got 6-fold upregulated in the UPEC strain UTI89 during infection [74]. This small RNA is found in widely studied UPEC strains CFT073 and UTI89 and in some extraintestinal pathogenic E. coli (ExPEC) strains but is missing from E. coli K-12 strains. PapR expression was shown to be regulated by Lrp as deletion of lrp in UTI89 abrogated the expression of papR.
Agglutination assays with yeast and human erythrocytes indicated that papR deletion increased mannose-resistant fimbrial expression, such as P-fimbriae, which was restored in the complemented strain. Similarly, adhesion assays using bladder and kidney epithelial cells showed increased mannose-resistant adhesion in UTI89ΔpapR, reinforcing PapR’s role in regulating fimbrial expression. Flow cytometry further confirmed that papR deletion doubled the fraction of P-fimbriated cells, while type-1 fimbriae levels remained unchanged. These findings suggest that PapR negatively regulates P-fimbrial expression at both the population and single-cell levels. To achieve this, PapR turns the pap-operon to the OFF state by interacting with papI, thereby preventing unnecessary fimbrial expression. While PapI promotes the ON state by interacting with the Lrp-DNA complex, PapR inhibits PapI by binding to a specific region (+74 to +96 nucleotides downstream of the papI translational start site) (Figure 1a). This likely facilitates RNase recruitment or modulates ribosomal loading to repress papI translation. Consequently, this inhibition disrupts the PapI-mediated translocation of the Lrp-DNA complex, thereby stabilizing the OFF phase [74].
In addition to PapR, the expression of P-fimbriae is subjected to epigenetic regulation by differential methylation of two GATC site within the 400 bp Pap regulatory region. Methylation at the distal GATC site maintains repression, whereas methylation at the proximal site promotes the ON phase by enabling PapI function [74] (Figure 1a). Stress-responsive small RNAs such as GcvB further modulate Lrp expression, while global regulators, including the cAMP receptor protein (CRP) and H-NS, impact P-fimbriae expression, with H-NS acting as a niche-specific repressor. The role of PapR extends beyond P-fimbriae silencing, as UPEC typically expresses only one fimbrial type at a time, leading to regulatory crosstalk [75]. The suppression of P-fimbriae by PapR may facilitate the compensatory upregulation of alternative adhesins, such as F1C fimbriae, ensuring adhesion plasticity. This suggests that PapR functions as a key regulatory checkpoint, restricting P-fimbriae expression to conditions where they are most beneficial for UPEC pathogenesis.
2.1.2. RyfA
In addition of PapR, P-fimbriae is regulated by another sRNA called RyfA. It is a 305-nucleotide sRNA implicated in oxidative and osmotic stress responses, biofilm formation, and swarming motility in Escherichia coli and Shigella dysenteriae [76, 77]. In Uropathogenic Escherichia coli (UPEC) strain CFT073, RyfA regulates genes involved in metabolism, respiration, and motility [76]. Interestingly, Shigella dysenteriae encodes two copies of RyfA namely RyfA1 and RyfA2 which are present in tandem in its genome and share about 95% in sequence identity [77]. RyfA1 but not RyfA2 was shown to be regulated by a second sRNA called RyfB1 and overproduction of RyfA1 disrupts the virulence-associated process of cell-to-cell spread and reduces the expression of ompC, a key outer membrane protein essential for Shigella pathogenesis.
RyfA is crucial for UPEC pathogenicity by regulating fimbrial expression, modulating fim genes for type 1 fimbriae and pap operons for P-fimbriae production. Additionally, ryfA mutants showed a 3.5-fold and 6.3-fold reduction in papA and papA_2 expression and 2.02 and 3.16-fold reduction papC and papC2, respectively confirming its role in pap operon regulation. Functional studies show that ryfA mutants exhibit significantly reduced fitness in a murine urinary tract infection (UTI) model, with a 124-fold decrease in bladder colonization and a 13-fold reduction in kidney colonization compared to the wild-type strain [76]. These findings suggest RyfA’s role in adhesion, and UPEC virulence.
2.1.3. LhrC
LhrC is a multicopy small RNA in Listeria monocytogenes that is activated in response to cell envelope stress and infection-relevant conditions [78, 79]. There are five variants of LhrC (LhrC1–5), each ranging from 111 to 114 nucleotides in length. While lhrC1–4 are clustered together in the genome, lhrC5 is located at a separate distant site. They are regulated by the stress-responsive two-component system (TCS) LisRK, linking their expression to environmental stressors such as low pH, ethanol exposure, and bile stress. These sRNAs play a critical role in virulence regulation by targeting genes involved in bacterial adhesion and immune evasion. Bioinformatics and β-galactosidase assays identified lapB mRNA, encoding a virulence-associated adhesin, as a direct target of LhrC1–5. Akin to PapR which negatively regulates the expression of P-fimbriae discussed above, LhrC negatively regulates lapB expression under bile and antibiotic stress or in blood, facilitating immune evasion by downregulating adhesin expression [78]. Unlike redundant regulatory mechanisms often seen in multicopy sRNAs, LhrC1–5 exhibit additive activity, with their CU-rich motifs binding to the AG sequence within the ribosomal binding site of target mRNAs, allowing precise post-transcriptional regulation. Transcriptomic and proteomic analyses further revealed that LhrC regulates additional virulence-associated genes, including the oligopeptide-binding protein oppA, the amino acid ABC transporter Lmo2349, and the CD4⁺ T-cell-stimulating antigen TcsA. Interestingly, LhrC variants demonstrated the ability to bind two oppA target mRNAs via two of their three CU-binding sites, suggesting a sophisticated regulatory mechanism [79]. These findings provide novel insights into LhrC-mediated gene regulation and highlight the potential of targeting LhrC for antimicrobial strategies aimed at disrupting Listeria virulence.
2.1.4. STnc640
STnc640 is a small RNA that plays a key role in adhesion and virulence regulation in Salmonella Enteritidis strain 50336. Sequence analysis revealed its shares 97% homology to its counterpart in Salmonella Typhimurium [80]. Using bioinformatics and RT-qPCR, Meng et al. demonstrated that STnc640 represses fimA, the major structural gene encoding type 1 fimbriae, via an Hfq-mediated post-transcriptional regulatory mechanism [57]. Unexpectedly, deletion of STnc640 enhanced bacterial adhesion to Caco-2 cells and increased virulence in a chicken infection model, suggesting that STnc640 acts as a negative regulator of adhesion-related genes. These findings imply that STnc640 may modulate virulence by fine-tuning bacterial adherence to host cells. Future studies, including overexpression experiments, are necessary to confirm its precise role in regulating fimbrial expression and virulence attenuation in Salmonella Enteritidis.
2.2. Invasion of the Host
A key attribute of successful pathogens is their ability to invade the host, facilitated by various virulence factors. For example, Escherichia coli (UPEC) produces α-hemolysin, encoded by the hlyCABD operon, which disrupts host cell membranes, causing lysis and tissue damage, thereby enhancing bacterial colonization of the urinary tract [81, 82]. Additionally, α-hemolysin modulates host immune responses by suppressing proinflammatory cytokine expression, allowing UPEC to evade immune detection. Similarly, Staphylococcus aureus secretes Protein A, which binds to the Fc region of antibodies, preventing opsonization and phagocytosis [83]. Bordetella pertussis produces adenylate cyclase toxin, increasing cyclic AMP levels in host cells and impairing immune function [84], while Pseudomonas aeruginosa releases Exotoxin A, which inhibits protein synthesis and induces immune cell death [85] (Table S2). Furthermore, Clostridium difficile produces Toxins A and B, which disrupt the cytoskeleton of intestinal cells, leading to inflammation and immune evasion [86]. These bacterial toxins employ diverse strategies to subvert host defenses, facilitating survival and pathogenesis. The following section also highlights the role of bacterial small RNAs in regulating virulence and host invasion, adding another layer of complexity to bacterial pathogenicity.Shiga toxin (Stx) is a major virulence factor of Shigella dysenteriae and enterohemorrhagic Escherichia coli (EHEC) [87]. It is an AB5 toxin, comprising an enzymatically active A subunit and a pentameric B subunit that binds globotriaosylceramide (Gb3) receptors on host cells. Upon internalization, the A subunit inhibits protein synthesis by cleaving an adenine residue from 28S rRNA, leading to cell death, epithelial damage, inflammation, and hemorrhagic colitis. In severe cases, Stx enters the bloodstream, causing endothelial damage in the kidneys and triggering hemolytic uremic syndrome (HUS), characterized by acute kidney injury, thrombocytopenia, and hemolytic anemia [88]. The toxin's genes, carried by lambdoid bacteriophages, enable horizontal transfer of virulence factors, driving bacterial evolution [89]. Stx is categorized into two primary groups, Stx1 and Stx2, with subtypes such as Stx1a, Stx2a, and Stx2d associated with severe disease.
2.2.1. StxS
Shiga toxin (Stx1) expression is regulated through three pathways: (1) the phage late promoter (PR'), activated by phage induction and the SOS response, integrates signals from quorum sensing, antibiotics, and inhibitors [90]; (2) a Fur-responsive promoter specific to Stx1, modulating expression based on iron and nitric oxide levels [91]; and (3) post-transcriptional regulation by small regulatory RNAs (sRNAs) [41]. During the lytic cycle, toxin genes are embedded within the late transcript of the phage and are regulated via antitermination of the PR′ late promoter. In the lysogenic state, transcription from PR′ is prematurely terminated at tR′, resulting in the production of a truncated 74nt long RNA, StxS as a byproduct of antitermination control. StxS represses Stx1 toxin production by binding to the ribosome binding site of stx1B, silencing its translation [41] (Figure 1b). Interestingly, StxS does not regulate stx2AB, which is exclusively induced during the lytic cycle, suggesting that other Hfq-dependent sRNAs may modulate Stx2a expression. This post-transcriptional regulation ensures minimal Stx1 production during lysogeny, acting as a safeguard. In addition to repressing Stx1, StxS activates the general stress response regulator RpoS by binding to its 5’ untranslated region (UTR), relieving repressive secondary structures and preventing Rho-dependent termination (Figure 1b). This activation enhances bacterial stationary-phase survival, increasing cell density under nutrient-limited conditions by approximately 20%. Another sRNA DicF, was shown to regulate Shiga toxin. As DicF responds to changes in oxygen level (the role of DicF is discussed in details in the bacterial stress response section), the pathogen can modulate the expression of the toxin by sensing the oxygen tension across different regions of the gut[92].
2.2.2. RNA III
Staphylococcus aureus α-hemolysin (Hla), a β-barrel pore-forming toxin, is essential to the pathogen's virulence [93]. Secreted as a monomer, Hla oligomerizes into a heptameric pore on host cell membranes, forming a 1–3 nm β-barrel pore that disrupts cellular ionic homeostasis and facilitates the passage of small molecules. At lytic concentrations, Hla causes cell lysis, while sublytic levels alter host signaling, affecting processes such as proliferation, inflammation, cytokine secretion, and cell-cell interactions [94]. Hla targets diverse human cell types, including epithelial, endothelial, and immune cells. Its activity is regulated by the agr quorum-sensing system and involves interactions with host lipids and the [95] receptor, contributing to species- and cell-type-specific effects [93]. Structural and mechanistic insights have informed therapeutic strategies aimed at mitigating Hla-mediated diseases.
The quorum-sensing system in S. aureus encodes two divergent RNAs: RNAIII, a central effector, and RNAII, encoding the agr operon [95]. RNAIII, 514 nt long sRNA upregulates Hla expression through two distinct mechanisms. First, it binds to the 5' untranslated region (UTR) of repressor of toxin (Rot) mRNA, a SarA ortholog that represses hla and other virulence factors, such as hlgB and hlgC (encoding gamma-hemolysin components) and sspB and sspC (encoding cysteine protease precursors) [96]. By silencing Rot, RNAIII promotes toxin and virulence factor expression. Second, RNAIII directly binds hla mRNA, exposing the Shine-Dalgarno sequence and enhancing translation [97].
Recent studies identified SprY, an sRNA encoded by the prophage φ12, as a novel virulence regulator through its interaction with RNAIII. SprY acts as an RNA sponge, binding the 13th stem-loop of RNAIII—the same site targeted by multiple RNAIII-regulated mRNAs, such as rot, spa, and eca [98]. This interaction prevents RNAIII from modulating its targets, enhancing Rot translation and reducing hemolytic activity. SprY is most active during early growth phases, facilitating colonization by suppressing toxin production. Conversely, SprX, another RNAIII-binding sRNA, exerts opposing effects, illustrating a complex network of sRNA interactions in fine-tuning virulence factor expression.
Additionally, Morales-Filloy et al. identified 5′-NAD-capped RNAs in S. aureus, including RNAIII, using NAD capture sequencing. RNAIII exhibits variable NAD modification levels [99]. NAD capping reduces α- and δ-hemolysin production and alters the expression of the splABCD operon, which is involved in tissue dissemination. Although in vitro studies suggest that NAD capping does not significantly affect RNAIII's secondary structure or translation, it may modulate regulatory functions in vivo by interacting with other factors. NAD capping is thought to fine-tune bacterial responses to redox state and quorum-sensing signals. NAD+/NADH 5′-modification of RNA occurs across bacteria, eukaryotes, and viruses, with RNA polymerases incorporating NAD during synthesis initiation. For example, in Escherichia coli, RNA NADylation is enhanced under low ATP conditions, weak ATP-RNA polymerase interactions, or DNA supercoiling [100]. Although NADylated RNA remains susceptible to RNase E processing and translation, it undergoes rapid degradation by NudC decapping, a process inhibited by the stringent response molecule ppGpp. This inhibition stabilizes NADylated RNAs during nutrient limitation, extending RNA half-life and facilitating efficient protein expression under stress conditions. In S. aureus, this mechanism may provide an energy-efficient strategy for ensuring stable toxin production and stress response under host-induced nutrient limitations.
2.2.3. BtsR1
Bacteria small RNA can also be managers of resource optimization ensuring the timely expression of their target only in presence of the stimuli as exemplified by the Bacillus thuringiensis strain YBT-1518. This strain harbors three nematicidal cry genes (cry55Aa, cry6Aa, and cry5Ba), with high toxicity against Caenorhabditis elegans [101]. The 51nt long sRNA BtsR1 directly regulates cry5Ba expression by binding to the ribosome-binding site (RBS) of cry5Ba mRNA, preventing translation. During free-living conditions, the suppression of cry5Ba by BtsR1 reduces toxin production, minimizing energy expenditure and avoiding host detection. Interestingly, once the bacterium is ingested by C. elegans, BtsR1-mediated silencing is alleviated, leading to the activation of Cry5Ba toxin expression within the host. The toxin then subverts the nematode's defenses and ultimately causes its death. This dynamic regulatory mechanism enables the bacterial pathogen to optimize toxin production in response to environmental conditions. Cry proteins are energetically costly, constituting over 25% of the bacterial dry mass, and their production during free-living conditions would waste resources. By silencing toxin synthesis via BtsR1, YBT-1518 avoids triggering host avoidance behaviors and ensures efficient ingestion by nematode hosts. Once inside the host, reduced transcription of BtsR1 allows the bacterium to activate Cry5Ba production, securing nutrients critical for its survival. This mechanism of toxin silencing highlights the ecological significance of sRNA-mediated regulation in balancing energy conservation with survival strategies. BtsR1 exemplifies how regulatory sRNAs enable pathogens to fine-tune gene expression in response to environmental and host-derived signals. This sRNA-mediated strategy reflects the co-evolutionary arms race between B. thuringiensis and nematode hosts [101].
2.3. Evading Immune Response
The small RNA which are discussed in the previous sections also contribute to growth of bacterial pathogens inside the host and at the same time equip the bacterial pathogen with defense to evade the host immune response. For example, RyhB and OxyS were known to regulate iron homeostasis and oxidative stress response to mitigate host immune defense and thus have emerged as major players of virulence in pathogenic strain of E. coli [102-104], Vibrio [105], Salmonella [62] and other gram-negative bacteria . Similarly,
Bacteria have evolved diverse strategies to evade the host immune response, enabling their survival and persistence within the host [11, 13]. One common approach is the modulation of surface structures to avoid recognition by the host's immune system. For instance, altering or masking pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharides or flagellin, prevents activation of Toll-like receptors (TLRs) on immune cells [106, 107]. Bacteria also secrete effector proteins through specialized secretion systems (e.g., Type III and Type IV secretion systems) to manipulate host cell signaling pathways, suppress pro-inflammatory responses, and inhibit apoptosis [58, 108] (Table S3). Additionally, some pathogens form biofilms—a protective extracellular matrix that shields them from phagocytosis and antimicrobial agents [109]. Intracellular pathogens like Salmonella and Listeria exploit host cells by residing in modified vacuoles or escaping into the cytosol, thus avoiding lysosomal degradation [110, 111]. There is growing evidence about the role of sRNA in immune evasion. For example, sRNAs modulate the expression of virulence factors that interfere with cytokine production, or inhibit oxidative stress pathways to enhance bacterial survival [112]. These multifaceted evasion tactics underscore the sophisticated interplay between bacteria and host immune defenses as discussed in the following examples.
2.3.1. FasX
Group A Streptococcus (GAS) such as Streptococcus pyogenes is a human-specific pathogen responsible for a broad spectrum of diseases including streptococcal pharyngitis (strep throat) to the highly invasive and often deadly necrotizing fasciitis (flesh-eating syndrome) [113, 114]. It produces a 205nt long sRNA, FasX that plays a major role in immune evasion of the pathogen. Its is regulated by the FasBCA system, with serotype-specific differences in basal expression potentially linked to single nucleotide polymorphisms (SNPs) in the fasX promoter. FasX reduces pilus expression by binding to different FCT-region of the mRNAs, inhibiting their translation [115] (Figure 1c). GAS pilus expression is tightly regulated, as it can enhance adhesion and biofilm formation but also increase bacterial susceptibility to neutrophil extracellular traps [115]. The interaction between FasX and its target pili gene is serotype specific. For example, in serotypes M1 and M2, FasX targets genes encoding the minor pilus protein (M1.cpa and M2.113), while in serotype M6 and M28, it regulates genes encoding the major pilus protein (M6.tee6 and M28.fctA). FasX also negatively regulates the expression of the fibronectin-binding proteins PrtF1 and PrtF2, which promote adhesion and internalization. FasX represses PrtF1/2 expression through posttranscriptional regulation by base-pairing with the prtF1 and prtF2 mRNAs within their 5′ untranslated regions, specifically overlapping the ribosome-binding sites. This duplex formation impedes ribosome access, thereby inhibiting mRNA translation. [43].
Conversely, FasX increases streptokinase (a plasminogen activator that produces serine protease plasmin which promotes bacterial metastasis) expression by stabilizing its mRNA [116]. These regulatory actions of FasX contribute to the pathogen's ability to cause disease by contributing to the transition from colonization to dissemination across multiple GAS serotypes as demonstrated in a bacteremia model using human plasminogen-expressing mice [116]. Reduced pilus expression decreases GAS adherence to host tissues, while increased streptokinase expression promotes tissue barrier breakdown and immune evasion (Figure 1d).
2.3.2. MTS1338
In Mycobacterium tuberculosis (Mtb), sRNA MTS1338 plays a crucial role in virulence and intracellular survival [37, 117]. MTS1338, highly conserved across pathogenic mycobacteria, is upregulated in dormant Mtb and macrophage-phagocytosed cells, suggesting a role in host-pathogen interactions. Elevated MTS1338 expression during infection is linked to transcriptional changes indicative of reduced metabolism and cell division, strategies associated with stress survival. Transcriptomic analyses revealed a characteristic response to MTS1338 overexpression during logarithmic and stationary phases, with increased resistance to low pH, reactive oxygen species (ROS), and reactive nitrogen species (RNS) in logarithmic-phase [118]. Survival analysis demonstrated enhanced stress resistance, highlighting MTS1338’s role in adapting to the macrophage environment. Interestingly, approximately 50% of differentially expressed genes (DEGs) in MTS1338-overexpressing strains overlapped with those activated in Mtb residing within macrophages, underscoring its importance in intracellular adaptation. These include mpt70, encoding an antigen involved in T-cell differentiation and expressed in later infection stages, Rv2034 and Rv2035, components of a toxin-antitoxin system and regulators of virulence and stress responses and cmtR (Rv1994c) and Rv2642, ArsR-family regulators linked to ROS detoxification and metal stress (Figure 1e).
MTS1338 is regulated by DosR, a transcriptional activator under hypoxic conditions, although it is not fully dependent on DosR [119, 120]. The DevR/DosR response regulator plays a crucial role in the virulence, dormancy adaptation, and antibiotic tolerance of Mycobacterium tuberculosis by regulating the dormancy regulon. Previously, we have developed recombinant tools to study gene regulation in Mycobacterium tuberculosis [121, 122]. Using this in vivo reporter assay, we characterized key residues involved in DevR–RNA polymerase interactions. In short, this system utilizes E. coli co-transformed with plasmids expressing M. tuberculosis RNA polymerase subunits (α, β, β′, and σA), a DevR expression cassette, and a GFP reporter under the DevR-regulated fdxA promoter. We found that the amino acid E280 in the α-subunit of RNAP and K513 and R515 in sigma factor A are involved in DevR-mediated transcription. In silico modeling suggests interactions between key residues of DevR (D172, E178) and sigma factor A (Q505, R515, K513) are required for DevR-mediated transcriptional regulation [123]. MTS1338 upregulation has been observed in hypoxic, acidic, and oxidative stress conditions, suggesting a role in dormancy. Biochemical studies indicate DosR binds with high affinity to MTS1338’s promoter region, enhancing its expression. Although PhoP does not directly bind to the MTS1338 promoter, it interacts with the DosR-MTS1338 complex, indicating a coordinated regulation mechanism. Taken together, these findings underscore MTS1338’s importance in M. tuberculosis pathogenicity, facilitating adaptation to macrophage-induced stress and promoting bacterial survival during chronic infection.
It is thus clear that bacteria have evolved a diverse repertoire of small RNAs (sRNAs) to regulate virulence factor expression, facilitating host infection and immune evasion. Beyond virulence regulation, sRNAs also contribute to complex stress response pathways, enabling bacterial adaptation to various host-induced stresses. The following section explores the role of sRNAs in mediating stress responses to counteract host defense mechanisms.
3. Role of Small RNA in Stress Response
A bacterial pathogen faces a multitude of hostile conditions that constitute significant stress within a host [30]. Immediately upon entry, the pathogen encounters the host's innate immune defenses, including antimicrobial peptides, reactive oxygen and nitrogen species produced by phagocytes. In addition, it is faced with host-mediated nutrient limitation (nutritional immunity). Furthermore, bacterial pathogens are subjected to further stresses like acidic pH within phagosomes, lysosomal enzymes, and continued oxidative and nitrosative attack [124]. Beyond direct immune assault, pathogens must also adapt to limited availability of essential nutrients like iron and specific carbon sources, competition with the host microbiota along with fluctuations in temperature and osmolarity [125, 126]. These diverse and dynamic stresses necessitate sophisticated bacterial adaptation mechanisms to ensure survival and successful infection.
Bacteria have evolved a diverse arsenal of defense mechanisms to counteract the stresses imposed by the host [30]. To combat reactive oxygen and nitrogen species (ROS/RNS), bacteria employ antioxidant enzymes like superoxide dismutase [127, 128], catalase [129], and alkyl hydroperoxide reductase [130]. To survive within acidic phagosomes, bacteria utilize acid tolerance systems, including proton pumps to maintain intracellular pH [131] and urease to generate ammonia [132]. Additionally, certain bacteria form biofilms, providing a protective matrix against immune cells and antimicrobial agents [109]. Furthermore, certain pathogens can modulate host immune responses directly, inhibiting phagosome maturation, escaping into the cytoplasm, or suppressing cytokine production, effectively manipulating the host's defenses to their advantage. Finally, nutrient limitation is addressed through metabolic flexibility, allowing bacteria to utilize alternative carbon sources and specialized scavenging and transport systems for essential nutrients like iron, [133]. Iron is a double-edged sword for living organisms. While indispensable for essential processes like respiration, DNA synthesis, and enzymatic catalysis, excess iron can be toxic due to the formation of reactive oxygen species (ROS) through the Fenton reaction. Therefore, maintaining iron homeostasis is critical for bacterial survival, especially within host environments where iron is often scarce due to host nutritional immunity. In E. coli, the transcription factor Ferric Uptake Regulator, Fur acts as a master regulator to control the iron uptake. In addition, Fur regulates a small RNA RyhB that equips the pathogen with stress response as outlined below.
3.1. RyhB
RyhB is a 95 nucleotide long sRNA that plays a key role in iron homeostasis and virulence in several pathogenic and non-pathogenic strains of bacteria including E. coli [134], Salmonella [135], and Vibrio [105] (Figure 2). It regulates targets through translational repression and mRNA degradation, often mediated by Hfq and RNase E. These include gene involved in TCA cycle and respiration and siderophore synthesis. RyhB is directly repressed by the transcriptional regulator called Ferric uptake regulator, Fur [136]. Under iron limiting conditions, RyhB was shown to play an iron-sparring response by inhibiting the translation of non-essential protein that uses Fe or Fe-S as a co-factor, thereby ensuring the essential proteins can ligate to Fe-containing co-factors to drive key metabolic processes needed for bacterial survival [136] (Figure 2a,b).
Previous works have suggested that deletion of fur did not impact virulence, but loss of RyhB or both Fur and RyhB impaired bladder colonization in the murine model of infection [102]. Although the role of Fur and RyhB have been well characterized in the non-pathogenic strains of E. coli, their contribution to the pathogenic strains remained elusive. To address this, using genomic and molecular biology tools like ChIP-seq, RNA-seq and Proteomics, we have characterized the Fur and RyhB regulon in uropathogenic strain of E. coli CFT073, revealing a complex interplay between iron acquisition, stress responses, and metabolic adaptation [136]. Fur directly and indirectly regulates a wide array of genes, including those involved in siderophore biosynthesis, iron uptake, and stress mitigation pathways, with RyhB modulating some components post-transcriptionally. Unique to CFT073, Fur-regulated pathways such as the siderophore aerobactin biosynthesis impose a metabolic burden leading to increasing amino acid demand, which may be mitigated by external amino acid uptake. We found that in the absence of Fur, a condition that mimics Fe-limiting environment, induced the RpoS mediated stringent stress response pathways in CFT073. This activation of the stringent stress pathways provides the pathogen with an anticipatory response that iron limitation could also be an indicative of other nutrient limitation as the pathogen often encounters in the nutrient limited urinary tract (Figure 2b). This regulatory network, distinct from the commensal strain MG1655, underscores the role of Fur and RyhB in optimizing iron and other nutrient acquisition and virulence in nutrient-limited host niches, offering insights into UPEC pathogenesis and potential targets for controlling infections [136]. Additionally, RyhB was also shown to play a key role in virulence of the avian pathogenic E. coli strain by regulating the expression of genes involved in biofilm formation, adhesion to brain microvascular endothelial cell line and contribute to meningitis development in the murine model of infection [103].
Recent work has identified the role of RyhB in persistence formation [137]. Persistors formation is one of the key strategies employed by the bacteria to convert itself into a metabolically inactive state to evade the host immune response (discussed in detail in the antibiotic section). Interestingly, Salmonella contains two copies RyhB in the genome-RyhB-1 and RyhB-2, which are encoded by separate genes found across all sequenced Salmonella species, including the distantly related Salmonella bongori [135]. These sRNAs share a conserved 33-base region, while the remainder of their sequences exhibit lower conservation. RyhB-1 and RyhB-2 have both overlapping and unique regulatory roles. As in E. coli, the expression of both ryhB-1 and ryhB-2 is repressed by the Fur; however, their activation is context-dependent. RyhB-1 is induced by oxidative stress caused by H₂O₂, whereas RyhB-2 is activated during the stationary growth phase. As in Salmonella, sp., Acuña et al have characterized two RyhB homologs, RyhB-1 and RyhB-2, in the fish pathogen Yersinia ruckeri [138]. Both homologs are induced under iron starvation, repressed by Fur, and stabilized by Hfq, similar to other Enterobacteriaceae. However, their functional roles differ. The ΔryhB-1 mutant exhibited increased motility, reduced biofilm formation, faster growth, and higher ATP levels, along with enhanced survival in salmon cell cultures. In contrast, the ΔryhB-2 mutant was non-motile and formed more biofilm. Gene expression analysis revealed that RyhB-1 negatively regulated multiple genes related to metabolism and motility, while RyhB-2 targeted only sdhC and bfr. These findings highlight the distinct, non-redundant roles of RyhB-1 and RyhB-2 in modulating Y. ruckeri physiology.
Interestingly, Coronel-Tellez et al have identified the small RNA IsrR that provides iron sparring response in Staphylococcus sp. akin to RyhB [139]. IsrR is repressed by iron, and it downregulates the expression of mRNAs encoding iron-containing enzymes, particularly those involved in anaerobic nitrate respiration. Structural analysis revealed three C-rich regions (CRRs) in IsrR, each contributing differently to target regulation. IsrR is essential for full virulence in a mouse septicemia model, highlighting its role in bacterial survival during infection. Although not orthologous to RyhB or other known sRNAs, IsrR regulates common targets, demonstrating convergent evolution in bacterial iron-sparing responses.
3.2. MsrI and Mcr11
MrsI (ncRv11846) is a 100 nucleotides long trans-encoded sRNA identified in Mycobacterium sp [140]. MrsI is regulated by the iron-dependent transcription factor IdeR, in both M. tuberculosis and M. smegmatis, driving its expression under iron-limiting conditions. High-throughput sequencing and differential expression studies identified MrsI as significantly upregulated during iron deprivation and membrane stress. Additionally, CRISPR-mediated knockdown of MrsI further confirmed its regulatory role in iron homeostasis, with 118 genes upregulated including bacterioferritin, hydrogenase maturation factor (HypF), and ferredoxin reductase (FprA). Functionally, MrsI represses bfrA mRNA, encoding bacterioferritin, an essential iron-storage protein [140]. This repression is mediated by a conserved six-nucleotide seed sequence and a seven-nucleotide apical loop. Collectively, these findings highlight MrsI as a critical regulator of iron metabolism, essential for Mtb adaptation to iron-limiting environments, akin to RyhB in Escherichia coli.
Another sRNA Mcr11, in Mycobacterium tuberculosis (Mtb), plays a crucial role in stress response during slow growth and chronic infection It is regulated by the transcription factor AbmR [141] by a mechanism that involves slowing down the transcribing RNA polymerase by binding to both RNAP and the DNA, similar to that observed for Rv1222 [142]. Bioinformatic predictions and qRT-PCR analyses identified Rv3282, fadA3, and lipB as direct targets of Mcr11, with base-pairing sites located immediately upstream of these genes. Notably, lipB expression was significantly de-repressed in Mcr11-deficient Mtb, confirming its regulatory role. Furthermore, Mcr11-mediated gene regulation was influenced by the presence of fatty acids, linking it to central metabolism. These findings highlight the importance of Mcr11 in Mtb’s adaptation and underscore the need to further explore sRNA stability and function in tuberculosis pathogenesis.
3.3. OxyS
OxyS is a 117-nucleotide long sRNA in Escherichia coli that plays a central role in the oxidative stress response [143]. Its expression is induced by the transcription factor OxyR under oxidative stress conditions. OxyS regulates the expression of approximately 40 genes [144] including genes involved in stress response, motility, DNA repair, and antibiotic resistance.One of the key functions of OxyS is to repress the transcriptional activator fhlA. Using NMR spectroscopy, SAXS, and molecular modeling, Štih et al. revealed that OxyS adopts a boomerang-like conformation consisting of 4 stem loops (SL1-SL4). The stem-loops SL1 and SL3 are positioned at opposite ends whereas SL4 shortens the unstructured linker to adopt a confirmation that allows repression of fhlA [145]. The chaperone protein Hfq enhances oxyS-fhlA interactions by destabilizing SL2 and partially unfolding SL4, promoting more efficient target binding, although OxyS retains regulatory function in the absence of Hfq, possibly forming stable Watson-Crick base pairs with fhlA, with magnesium ions facilitating partial unfolding for enhanced base pairing. This structural plasticity likely underlies OxyS’s ability to regulate multiple targets. OxyS also negatively regulates the expression of the alternative sigma factor RpoS, which is essential for general stress resistance in E. coli. While initial studies proposed direct base pairing between oxyS mRNA and the rpoS leader sequence, later research demonstrated that OxyS has low affinity for the rpoS mRNA leader region despite its strong binding to Hfq [146]. This suggests an indirect mechanism, where OxyS sequesters Hfq, reducing its availability to other sRNAs, including DsrA, a known activator of rpoS.
Overexpression of OxyS inhibits swimming and swarming motility and reduces curli expression, linking it to biofilm formation [40]. Additionally, OxyS contributes to cellular stress adaptation by inducing temporary cell cycle arrest, allowing for DNA repair [147]. To achieve this, it represses the expression of transcription termination factor NusG, leading to the activation of the cryptic prophage gene kilR. KilR inhibits FtsZ, a key protein in bacterial cell division, slowing proliferation and allowing DNA repair to proceed effectively (Figure 2c). OxyS influences antibiotic susceptibility, particularly cephalothin resistance, by modulating target gene expression [148]. Transcriptomic and phenotypic analyses by Cho et al. identified 27 differentially expressed OxyS-regulated genes, including cycA (D-serine/alanine/glycine:H+ symporter) and cysH (phosphoadenosine phosphosulfate reductase). Knocking out 17 genes out of the 27 OxyS targets exhibited increased resistance. 9 of were linked to the cAMP receptor protein (CRP), a global transcriptional regulator in E. coli. These findings suggest that OxyS modulates multiple pathways to confer stress response and may serve as a molecular marker or target for identifying and combating antibiotic resistant strains.
3.4. DsrA
There are two global regulators that plays a key role in stress response in E. coli. The first is the stress induced sigma factor RpoS that equips the pathogen with necessary adaptations to cope up with the stress. The second is H-NS, a nucleoid-associated protein in bacteria akin to histones in the eukaryotic systems that binds across the genome and acts as a global transcriptional repressor, silencing genes involved in stress responses and virulence. Although several transcription factors regulate the expression of RpoS and H-NS, an 87-nucleotide long sRNA called DsrA regulates the expression of both RpoS and H-NS antagonistically. DsrA activates the expression of rpoS posttranscriptional by binding to binds to 5’UTR and disrupting inhibitory secondary structures of rpoS transcript [149]. Alternatively, it represses the expression of hns causing derepression of H-NS silenced gens that are involved in stress response.
DsrA promotes acid resistance by activating genes involved in acid stress response including hdeAB, gadAX, and gadBC operons in E. coli K-12 strains, as well as pathogenic in E. coli O157:H7, contributing to bacterial endurance in acidic environments such as the stomach [150]. In Salmonella enterica serovar Typhimurium, DsrA influences central carbon metabolism (CCM) and NAD(H) homeostasis by altering expression of genes involved in glycolysis, pyruvate metabolism, the TCA cycle, and NADH-dependent respiration. GFP-fluorescence based reporter assay identified pflB, encoding pyruvate-formate lyase, as a direct target of DsrA repression. DsrA base pairs with pflB mRNA, recruiting RNase E to degrade it, thereby reducing pflB stability. This repression lowers the NAD+/NADH ratio, helping maintain redox balance [151]. Furthermore, Additionally, DsrA, plays a key role in prophage λ induction by relieving repression of rcsA through antagonizing H-NS. This leads to increased capsular polysaccharide (cps) expression and RecA-independent prophage induction. Unlike RcsA, which requires RcsB for induction, DsrA functions independently of RcsB in stationary phase, suggesting an alternative pathway for prophage activation [152, 153]. Recent findings suggest DsrA also plays a role in replication initiation, requiring Hfq, Dps, and OxyR [154].
3.5. RybB
The outer membrane (OM) is crucial for the survival of Gram-negative bacteria. In Escherichia coli, the Sigma Factor E, SigE plays a crucial role in envelop stress response by detecting signals that are indicative of OM dysfunction and thereby initiates an adaptive response by upregulating genes involved in the biogenesis, transport, and assembly of key OM components, including lipopolysaccharides (LPS), phospholipids, and outer membrane proteins (OMPs) [155]. Additionally, sigE regulates the expression of proteases and chaperones that contribute to OM maintenance and repair, ensuring membrane integrity under envelop stress conditions. One key player in this stress response is a 79-nucleotide long sRNA called RybB. The expression of RybB is regulated by SigE and the stress-associated alarmone ppGpp in Escherichia coli and Salmonella sp [156]. Like other trans-acting sRNAs, RybB’s function depends on RNA chaperones, including Hfq and ProQ. Hfq stabilizes RybB and facilitates its interactions with target mRNAs, while ProQ protects it from degradation. It's key targets include mRNAs encoding outer membrane proteins such as ompC, ompW, ompA, and fadL [157] (Figure 2d). By targeting these mRNAs, RybB reduces the production of porins and fatty acid transporters, facilitating rapid adjustments to envelope composition. In Salmonella, RybB plays a greater role to cope up with envelop stress by regulating additional targets like ompD, ompF, ompN, ompS, and tsx. This ability to modulate OMP expression is crucial for bacterial adaptation to stressors, including changes in osmolarity, pH, and exposure to antimicrobial agents. Interestingly Adams et al. identified FtsO, an ORF-internal sRNA derived from the ftsI mRNA, which plays a crucial role in bacterial cell division acts a RNA sponge to negatively regulate RybB. Since ftsI encodes a penicillin-binding protein essential for cell division, the regulation of RybB by FtsO suggests a mechanism linking membrane stress response with the division cycle [158]. RybB also acts as a negative regulator of biofilm formation by negatively regulating the expression of Curlin subunit gene D, CsgD. Beyond biofilm regulation, RybB promotes stationary-phase cell lysis, a process critical for population turnover, eliminating damaged cells, and preventing mutation accumulation. Disrupting RybB or MicA impairs lysis, reducing survival and increasing mutation rates, ultimately causing population collapse.
In summary, bacterial small RNAs play a pivotal role in enabling pathogens to withstand various host-induced stresses, thereby contributing significantly to bacterial pathogenesis and host-pathogen interactions. Moreover, bacteria frequently encounter antibiotics, which are administered to eliminate pathogenic infections. The following section explores the role of small RNAs in mitigating the adverse effects of antibiotic exposure and enhancing bacterial survival.
4. Role of Small RNA in Antibiotic Resistance
As discussed in the previous sections, bacterial pathogens pose a significant global threat due to their diverse virulence factors and stress-responsive mechanisms, which enable them to evade host immunity and establish infections. To combat these pathogens, antibiotics were developed as a crucial means of treatment [159]. However, in the recent years the efficacy of the antibiotics have gone down due to the rise of multidrug-resistant strains [18]. Antibiotics enter bacterial cells through various mechanisms, depending on their class and the bacterial cell membrane/wall structure and targets essential processes thereby killing the pathogen. For instance, Beta-lactams (e.g., penicillins, cephalosporins) diffuse through the peptidoglycan layer in Gram-positive bacteria, inhibiting cell wall synthesis [160] whereas aminoglycosides (e.g., gentamicin, streptomycin) pass through porin channels in Gram-negative bacteria and bind ribosomal RNA, disrupting protein synthesis [161]. Similarly, Tetracyclines (e.g., doxycycline) utilize passive diffusion and active transport to inhibit ribosomal function [162]. Macrolides (e.g., erythromycin, azithromycin) diffuse through membranes to bind the 50S ribosomal subunit, halting protein synthesis [163]. Fluoroquinolones (e.g., ciprofloxacin) enter via porins and inhibit DNAA replication by targeting DNA gyrase and topoisomerase IV [164]. Sulfonamides (e.g., sulfamethoxazole) mimic para-aminobenzoic acid (PABA) to block folic acid synthesis [165]. Glycopeptides (e.g., vancomycin) target cell wall precursors, preventing synthesis [166]. Taken together, each class of antibiotics has a unique mechanism of entry and action, which is crucial for their effectiveness and the development of resistance mechanisms.
To curb the effect to antibiotics, bacteria have utilized small RNAs to developed elaborate strategies that I have discussed in the following section. For example, treatment of last-line antibiotics, including linezolid, ceftobiprole, tigecycline, and vancomycin to methicillin-resistant Staphylococcus aureus (MRSA) exhibited a coordinated sRNA response [167]. Similar sRNA dependent response to sub-MIC levels of eight clinically relevant antibiotics and tigecycline was also observed in DOT-T1E isolate of Pseudomonas putida [168] and Salmonella Typhimurium respectively. All these studies underscore the importance of small RNA in both antibiotic resistance as well as susceptibility making them novel targets for drug development. To understand the role of small RNA in antibiotic resistance and susceptibility, I have divided into following 3 sections based on the strategies the multi-drug-resistant bacteria employ to curb the effects of the antibiotics.
4.1. Inhibiting Drug-Uptake Systems
As detailed in the previous paragraphs, antibiotics enter bacterial cells through various mechanisms, ranging from simple diffusion to energy-dependent transport. To hinder antibiotic entry, bacteria employ sRNAs that target membrane proteins involved in antibiotic uptake, as discussed in the following section.
4.1.1. GcvB
One of the key players of antibiotic resistance in E. coli and Vibrio is the small RNA GcvB, a 205 nucleotides long noncoding sRNA, transcribed divergently from the gcvA gene, the latter encodes for the transcriptional activator of the glycine cleavage (GCV) pathway. In presence of glycine, the transcription of gcvB is activated by GcvA and GcvR. Although GcvB was initially associated with acid stress response [169], recent studies have elucidated its role in antibiotic resistance. Deleting gcvB in E. coli leads to heighten sensitivity to various aminoglycoside antibiotics, including neomycin, streptomycin, kanamycin, kasugamycin, and spectinomycin, even at low concentrations [170]. GcvB consists of 5 stem-loops (SL1-5) with three conserved seed sequences, R1, R2, and R3, to regulate multiple target genes [171]. The G/U-rich R1 seed sequence located between SL1 and SL2 can base-pair with the majority of target mRNAs. Although the seed sequence R2 primarily repress cycA mRNA in E. coli and Salmonella whereas the R3 seed sequence, located in SL4, regulates several mRNAs, including global transcriptional regulators PhoP, Lrp, and SroC [172, 173] (Figure 3a).
GcvB contributes to antibiotic resistance in Escherichia coli by post-transcriptionally downregulating the expression of the inner membrane protein CycA. In addition to its role in amino acid (D-glycine, D-serine, D-alanine) import, CycA facilitates the uptake of aminoglycoside antibiotics such as cycloserine. GcvB mediates this regulation by base-pairing with cycA mRNA, promoting its degradation and thereby reducing CycA protein levels, ultimately limiting cycloserine entry into the cell [174]. Two regions of GcvB, spanning nucleotides +124 to +161 and +73 to +82, exhibit complementarity to the cycA mRNA. Transcriptional fusions of cycA to lacZ revealed that this cycA mRNA region is essential for GcvB-mediated regulation [175]. Muto et al. deciphered that either of the abovementioned complementary region of GcvB can independently base-pair with and repress cycA-lacZ, with significant repression loss occurring only when both regions are mutated [170]. Additionally, mutations in the cycA gene can lead to increased resistance to DCS by reducing its uptake into the bacterial cell. Moreover, GcvB plays a role in promoting mutagenic break repair (MBR) by enabling a robust rpoS response while suppressing the competing sigE (membrane stress) response. gcvB deletion impairs MBR and rpoS-dependent transcription without reducing rpoS protein levels. Restoring rpoS levels or inhibiting sigE response induction rescues these defects, suggesting that GcvB maintains membrane integrity resulting in decreased sigE activation. This balance prevents sigE from outcompeting rpoS for RNA polymerase, thereby supporting mutagenesis.[176]. Furthermore, using transcriptomics and proteomics-based approach, Liu et al have identified the role of GcvB in regulating type IIII secretion system in the marine bacterium Vibrio alginolyticus thereby expanding its role in bacterial virulence [171].
4.1.2. MicF
The micF RNA is a 93-nucleotide long sRNA that plays a significant role in the regulation of antibiotic resistance in Escherichia coli. It primarily functions by binding to the mRNA of the outer membrane porin protein OmpF, preventing its translation [49, 177]. OmpF forms trimeric channels that allow the passive diffusion of small hydrophilic molecules, including nutrients and antibiotics, across the outer membrane [178]. By repressing OmpF production, micF reduces the permeability of the bacterial cell membrane to antibiotics, thereby decreasing the intracellular concentration of these drugs and enhancing bacterial survival. The expression of micF is tightly regulated by several global stress response regulators, such as MarA, Rob, and SoxS [177] (Figure 3a). These regulators are activated under various stress conditions, including the presence of antibiotics, and oxidative stress conditions. When activated, they induce the expression of micF, leading to a rapid decrease in OmpF levels and an increase in antibiotic resistance.
Carrier et al. recently elucidated the role of MicF in the activation of the outer membrane porin OppA [179]. OppA plays a key in the survival of uropathogenic E. coli by importing peptides allowing the bacteria to thrive in otherwise nutrient limited and hostile urinary tract [133] . Additionally, OppA has also been implicated in antibiotic sensitivity in UPEC, particularly in response to Polymyxin B [180]. The uptake of certain toxic compounds, including the synthetic tripeptide tri-L-ornithine [107] and the novel antibiotic GE81112 [108], is also dependent on OppA. Under standard growth conditions, GcvB and Hfq inhibit ribosomal binding to the Shine-Dalgarno sequence, resulting in low oppA expression. However, under membrane stress, MicF is activated and binds to the 5’ untranslated region (UTR) of the oppA transcript, enhancing translation and leading to increased oppA expression.
4.1.3. EsrA and Sr0161
Pseudomonas aeruginosa is a prominent opportunistic human pathogen, responsible for approximately 3,000 deaths annually in the United States [181]. Multidrug-resistant (MDR) P. aeruginosa infections are often treated with carbapenem antibiotics like meropenem; however, 2–3% of clinical isolates produce carbapenemase, rendering them resistant to this antibiotic class. Carbapenem uptake in P. aeruginosa is mediated by the major porins OprD and OpdP, with oprD deletion increasing carbapenem resistance in MDR isolates [182].
Using Hi-GRIL-seq, Sr0161 and ErsA were identified as sRNAs that negatively regulate oprD expression via interactions with its 5′ untranslated region (5′-UTR) [183] (Figure 3a). Overexpression of Sr0161 enhances resistance to meropenem [183], while deletion of sr0161 or ersA significantly increases susceptibility to this antibiotic. The RNA chaperone Hfq also contributes to carbapenem resistance by regulating oprD and opdP porins, as shown by the increased carbapenem susceptibility observed in hfq deletion mutants [184].
4.2. Modification of Cell envelope
Lipopolysaccharides (LPS) are crucial components of the outer membrane of Gram-negative bacteria, playing key roles in maintaining membrane integrity, stress response, and virulence [185]. LPS consists of three main parts: lipid A, a core oligosaccharide, and an O-antigen. The lipid A portion anchors LPS to the membrane and is a target for polymyxins class of antibiotics. such as colistin [186]. However, modifications to LPS, such as the addition of phosphoethanolamine which leads change in the charge and structure of the lipid A can reduce susceptibility to polymyxins. Polymyxin resistance in Escherichia coli is mediated by the enzyme EptB, which modifies lipopolysaccharide (LPS) with phosphoethanolamine [180]. The translation of eptB mRNA is inhibited by the small RNA (sRNA) MgrR, which is itself suppressed by base pairing with the sponge sRNA SroC. As a result, the loss of MgrR enhances, while the loss of SroC reduces, resistance to polymyxin B. Additionally, eptB expression is negatively regulated by the sRNA ArcZ, whose levels are governed by the ArcA–ArcB two-component system, which senses aerobic and anaerobic conditions [187]. Interestingly, the deletion of Hfq—a protein essential for the activity of these sRNAs results in increased susceptibility to polymyxin B. This phenomenon may be attributed to a compromised cell envelope stress response mediated by the sigma factor RpoE. RpoE not only promotes eptB transcription but also regulates other Hfq-dependent sRNAs involved in LPS biogenesis and modification. The following section discusses the role of 2 small RNA that provides antibiotic resistance by modification of bacterial cell membrane.
4.2.1. SprX (Modification of Cell Membrane)
Staphylococcus aureus is a major cause of bacteremia, infective endocarditis, and osteomyelitis, and frequently colonizes medical implants [188]. The emergence of methicillin-resistant S. aureus (MRSA) strains has severely limited treatment options, often leaving glycopeptide antimicrobials, which target peptidoglycan cell-wall synthesis, as the primary therapeutic option [189]. The sRNA SprX encoded within the pathogenicity island, is highly conserved among Staphylococcus aureus strains and present in several S. aureus phages [190]. Most strains carry a single copy, while the Newman strain has three copies with minor nucleotide differences. SprX plays a pivotal role in modulating antibiotic resistance mechanisms by directly base pairing with the translation initiation site of spoVG mRNA, a key regulator of capsule production, virulence, and cell wall metabolism. This interaction prevents ribosome loading, thereby suppressing SpoVG protein expression [198]. SpoVG regulates critical pathways associated with methicillin and glycopeptide resistance, including activation of the lytSR operon, the glycine glycyltransferases (femAB) which mediate peptidoglycan crosslinking and repression of the murein hydrolase lytN [199]. By inhibiting SpoVG, SprX increases MRSA susceptibility to glycopeptides. In addition, SprX controls virulence factors such as delta-haemolysin and clumping factor B by interacting with hld and clfB transcripts, while also influencing IsaA expression and biofilm formation [191]. In S. aureus HG001, it plays a role in modulating glycopeptide resistance and complement-binding protein [192, 193]. Furthermore, SprX contributes to antibiotic resistance by regulating autolysin production through its interaction with walR mRNA [194].
4.2.2. Sr006
The sRNA Sr006, transcribed from the intergenic region between 182,570 and 182,693 bp on the minus strand, was identified in chimeric RNA molecules with lipid A diacylation enzyme pagL mRNA[195] and functions as a positive post-transcriptional regulator of pagL in Pseudomonas aeruginosa virulence and host adaptation, as revealed by Hi-GRIL-seq. Western blot and RT-qPCR analyses demonstrated that overexpression of Sr006 significantly increased pagL mRNA and PagL protein levels, while deletion of Sr006 had no effect. Functional studies revealed that Sr006-mediated upregulation of PagL resulted in enhanced deacylation of lipid A, shifting it to a less pro-inflammatory form, as confirmed by MALDI-MS and GC-FID analyses. Furthermore, Sr006-dependent regulation of pagL increased polymyxin B resistance in P. aeruginosa strains lacking LPS aminoarabinose modifications. Unlike other sRNA-mediated regulatory mechanisms requiring the RNA chaperone Hfq, Sr006’s regulation of pagL was Hfq-independent, suggesting an alternative mechanism of action. This regulatory pathway may enable P. aeruginosa to adapt to the host environment, potentially through modulation of Sr006 expression in response to host interactions.
4.3. Activation of Efflux Pumps
The tripartite efflux pump systems in E. coli—AcrAB-TolC, MacAB-TolC, MdtABC-TolC, and EmrAB-TolC plays a crucial roles in multidrug resistance by expelling a variety of toxic compounds [196] (Figure 3a). The AcrAB-TolC system consists of AcrA, a periplasmic adapter protein that bridges the inner and outer membranes, AcrB, a Resistance-Nodulation-Division (RND) transporter responsible for recognizing diverse substrates such as antibiotics and bile salts, and TolC, an outer membrane channel [197]. The acrAB genes are organized within an operon, while tolC is located separately on the E. coli chromosome within an operon that includes ygiB and ygiC. Both the acrAB and tolC promoters are regulated by the MarA, SoxS, and Rob transcriptional regulators and are upregulated in response to superoxide stress and exposure to heavy metals, bile salts, and antibiotics, reflecting the efflux pump's role in exporting these harmful substances from the cell. [198] (Figure 3a). In addition to AcrAB-TolC efflux pump, TolC was found associated with MacA-MacB and MtdA-MdtB-MdtC to form MacAB-TolC [199] and MdtABC-TolC efflux pumps respectively [200]. The MacAB-TolC pump exports macrolides like erythromycin and secretes virulence factors, whereas the MdtABC-TolC system confers resistance to bile salts and heavy metals, aiding bacterial colonization in the gut. Lastly, the EmrAB-TolC system, part of the Major Facilitator Superfamily (MFS), uses a proton/drug antiporter mechanism to resist hydrophobic antibiotics [201] (Figure 3a). Collectively, these efflux pumps are vital for bacterial survival in hostile environments and contribute significantly to antibiotic resistance. Recent works have deciphered the regulations of these multi—drug efflux pumps by small RNAs as discussed in the following paragraphs.
4.3.1. CsiR
Strong evidence indicates that antibiotics are a major driving force behind bacterial resistance [202]. Ciprofloxacin, a third-generation synthetic quinolone, is widely used in clinical and agricultural settings, exerting its antibacterial effects by inhibiting bacterial DNA helicase and effectively targeting Pseudomonas aeruginosa, Escherichia coli, Proteus vulgaris, and other Enterobacteriaceae [203]. CsiR, a non-coding RNA, functions as a negative regulator of emrB, a multidrug efflux pump gene that plays a crucial role in ciprofloxacin resistance in Proteus vulgaris strain P3M. Gene expression analysis revealed that emrB was significantly upregulated under ciprofloxacin treatment. Interestingly emrB expression was even higher in a ∆csiR mutant strain, confirming that CsiR inhibits emrB expression, thereby decreasing ciprofloxacin efflux and reducing bacterial resistance. Bioinformatics predictions identified a specific binding site for CsiR in the 5′ untranslated region (UTR), near the Shine-Dalgarno (SD) sequence of emrB mRNA, suggesting a direct post-transcriptional regulatory interaction. This was experimentally validated through microscale thermophoresis (MST) assays, which showed a strong binding affinity between wild-type CsiR and emrB mRNA, with a dissociation constant of 11.5 ± 3.56 μM. Mutation of the predicted emrB binding site in CsiR resulted in abolishing its interaction with emrB. Furthermore, in vivo studies confirmed that mutating this binding site in CsiR led to a loss of repression, restoring emrB expression to levels observed in the ∆csiR strain and significantly increasing bacterial survival in presence of ciprofloxacin. These findings strongly suggest that CsiR-mediated repression of emrB represents a key regulatory mechanism controlling antibiotic resistance in Proteus vulgaris highlighting the potential of targeting ncRNA-efflux pump interactions for antimicrobial therapy[204]. The absence of CsiR also affected resistance to polymyxin B and erythromycin, indicating its multifunctional role in antibiotic resistance.
4.3.2. PA0805.1
Pseudomonas aeruginosa initiates swarming motility in response to semisolid surfaces and nitrogen sources relevant to the human lung. Although the transcriptional regulation of swarming has been well studied, posttranscriptional regulation by small noncoding RNAs (sRNAs) remains underexplored. Coleman et al. found that the sRNA PA0805.1 was upregulated 5-fold under swarming conditions. Overexpression of PA0805.1 in wild-type (WT) PAO1 led to reduced swarming, swimming, and twitching motilities, along with increased adherence, cytotoxicity, and tobramycin resistance, while its deletion (ΔPA0805.1) increased tobramycin susceptibility [205].
Transcriptomic and proteomic analyses of the PA0805.1-overexpressing strain revealed significant alterations in gene and protein expression, with 1,121 differentially expressed genes and 258 proteins. Among these were 106 transcriptional regulators, two-component systems, and sigma/anti-sigma factors implicated in antimicrobial resistance and virulence. Notably, the upregulation of multidrug efflux systems mexXY and mexGHI-opmD provides a mechanistic basis for increased tobramycin resistance (Figure 3a). Additionally, ClpD, a protease essential for biofilm dispersal and virulence, was upregulated, potentially contributing to adaptive resistance.
Further regulatory changes included downregulation of type IV pilus genes and transcription factors such as PilGH and ExsD, the latter being a negative regulator of the type III secretion system (T3SS). In contrast, key regulators involved in virulence, quorum sensing, and biofilm formation—AlgR, LasR, RhlR, MvaT, and PsrA—were differentially expressed, reinforcing a shift towards an antibiotic-resistant and persistent phenotype [205]. These findings underscore the role of PA0805.1 in modulating antibiotic resistance pathways in P. aeruginosa, particularly through efflux system activation and biofilm-associated mechanisms, highlighting its potential as a target for therapeutic intervention.
4.3.3. SdsR and RyeA
Small RNAs (sRNAs) SdsR and RyeA in Escherichia coli, transcribed from opposite strands of the same genomic locus, exemplify a unique sRNA-mediated toxin-antitoxin (T/A) system [206].. Ectopic expression of SdsR during exponential growth caused Hfq-dependent cell death and filamentation, with RNA-seq analysis revealing altered expression of 209 genes, including the repression of yhcB, an inner membrane protein critical for survival. Overexpression of RyeA mitigated SdsR-induced toxicity, highlighting an antagonistic interplay between the two sRNAs. These findings uncover a novel regulatory role for SdsR in bacterial stress responses and emphasize its functional relationship with RyeA in maintaining cellular homeostasis [206].
Importantly, SdsR is essential for the posttranscriptional regulation of the AcrAB-TolC efflux system, directly base-pairing upstream of the tolC ribosomal binding site to repress its expression, thereby reducing TolC protein production [207]. This repression leads to increased susceptibility to antibiotics such as novobiocin indicating that SdsR negatively impacts the bacterium’s ability to expel toxic compounds. Similarly, overexpression of SdsR has been shown to heighten susceptibility to quinolone antibiotics, including levofloxacin, nalidixic acid, and norfloxacin, in multidrug-resistant strains of E. coli and Salmonella.
Additionally, SdsR has been linked to broader stress responses and genetic regulation SdsR has been linked to broader stress responses and genetic regulation For instance, SdsR appears to influence bacterial mutagenesis, as its interaction with mutS has been associated with an increased mutation rate under subinhibitory ampicillin treatment [208]. This suggests a potential role for SdsR in adaptive stress responses. Furthermore, SdsR overexpression was found to reduce biofilm formation in E. coli, suggesting that it may influence bacterial surface attachment and persistence. These findings collectively highlight SdsR as a key modulator of antibiotic susceptibility and stress adaptation, though its precise physiological roles warrant further investigation
4.4. Biofilm-Mediated Increased Drug Resistance in Bacterial Pathogens
One of the adaptive strategies employed by bacteria to evade the host immune response and antibiotic action is biofilm formation [209-211]. Biofilms are structured communities of bacteria, encased in an extracellular polymeric substance (EPS) composed of polysaccharides, proteins, lipids, and extracellular DNA [212]. This EPS matrix acts as a physical barrier, limiting antibiotic penetration and creating chemical gradients that reduce the effectiveness of antibiotics [213]. The formation of biofilms is tightly regulated by transcriptional factors. In Pseudomonas aeruginosa, key regulators include AlgR, which promotes the production of alginate (a major biofilm polysaccharide), and the two-component systems GacS/GacA and BfiSR, which regulate biofilm maturation [214]. The quorum-sensing regulators LasR and RhlR also play crucial roles in coordinating biofilm development by inducing EPS production and virulence factor expression [215].
sRNAs are now recognized for their crucial role in biofilm formation, a prevalent multicellular bacterial lifestyle across various bacterial species [39, 216]. In Escherichia coli, CsrB and CsrC sequester CsrA, results in increased expression of the genes involved in extracellular polysaccharide synthesis. Similarly, in Salmonella enterica, small RNAs ArcZ and SdsR regulate the transcriptional switch CsgD, which governs biofilm matrix production and motility repression. Additional sRNAs, McaS, RprA, and OmrA/OmrB, integrate environmental signals to modulate csgD expression, thereby influencing lifestyle transitions. The second messenger c-di-GMP further reinforces biofilm formation by promoting extracellular polysaccharide production. [217]. Moreover, in Vibrio cholerae, the sRNA RybB represses ompA, modulating membrane permeability to favor biofilm formation and environmental adaptation [105]. Finally, in Bacillus subtilis, BsrG and SR1 contribute to biofilm formation, with SR1 modulating metabolic pathways for extracellular polysaccharide synthesis and BsrG enhancing stress adaptation. Collectively, these regulatory sRNAs enable bacteria to fine-tune biofilm formation in response to environmental cues.
Although small RNAs (sRNAs) play a crucial role in regulating biofilm formation across multiple bacterial pathogens, this review focuses on three specific sRNAs involved in biofilm formation in Pseudomonas sp. The recent rise of multi-drug resistance (MDR) in Pseudomonas aeruginosa has become a significant global health concern, largely driven by the overuse and misuse of antibiotics in clinical and environmental settings. Biofilm formation is a key contributor to this multi-drug-resistant phenotype, enhancing bacterial survival and persistence. A deeper understanding of the molecular mechanisms underlying biofilm formation will provide new avenues for the development of targeted therapeutic strategies.
4.4.1. AS1974
The 127 nucleotide long sRNA AS1974 has been implicated in biofilm-associated antibiotic resistance in Pseudomonas sp. Hfq RIP-seq analysis identified AS1974 as being downregulated in MDR P. aeruginosa isolates, with its overexpression sensitizing cells to aminoglycosides, cephalosporins, and carbapenems [218, 219] suggesting AS1974 as a negative regulator of antimicrobial resistance. This is achieved by AS1974 mediated repression of ndvB, a gene encoding a biofilm-associated protein required for the synthesis of periplasmic glucans that sequester antibiotics and preventing its entry into the bacterial cell. Deletion of ndvB sensitizes biofilms to gentamicin and ciprofloxacin [219]. Transcriptomic analysis revealed that AS1974 sRNA plays a regulatory role in multiple drug resistance pathways by downregulating key resistance genes, including mexD- a component of multi-drug efflux system MexC-MexD-OprJ, prc, periplasmic tail specific protease, an enzyme involved in processing off C-terminal region of penicillin binding protein 3 andd chtA- a TonB dependent siderophore receptor (Figure 3b).
To investigate the regulation of AS1974, researchers analyzed its promoter region in drug-susceptible and multidrug-resistant (MDR) strains. Chromatin immunoprecipitation followed by qPCR (ChIP-qPCR) revealed stronger RNA polymerase binding to the AS1974 promoter in susceptible strains, hinted toward a possible epigenetic regulation of AS1974 that downregulates its transcription in resistant strains akin methylation-mediated regulation of P-fimbriae (discussed in Section 2.1.1). Bisulfite genomic sequencing identified three methylation sites upstream of the AS1974 promoter in resistant strains but are missing from susceptible strains. These sites were located at –16, –66, and –73 relative to the transcriptional start site (TSS) of AS1974 (Figure 3b). A GFP reporter assay demonstrated that AS1974 transcription was suppressed in resistant strains, confirming a functional impact of methylation. These results suggest that promoter methylation inhibits AS1974 transcription by restricting RNA polymerase binding, contributing to drug resistance in MDR strains [219].
4.4.2. PhoS
PhoS as a 66-nucleotide long sRNA that is encoded at the 3’UTR of PhoR, although the expression of PhoS is driven by its own promoter (Figure 3b). PhoR is a sensor kinase that regulates bacterial responses to phosphate availability by toggling between kinase and phosphatase modes. Under phosphate-limiting conditions, PhoR functions as a kinase, phosphorylating its cognate response regulators, PhoB or PhoP, which then activate expression of genes involved in adaptation to phosphate scarcity. Conversely, under phosphate-replete conditions, PhoR assumes a phosphatase role, maintaining PhoB/PhoP in an inactive, dephosphorylated state (Figure 3c).
Studies in E. coli and B. subtilis have identified two mechanisms regulating PhoR activity. In E. coli, the predominant pathway involves the PstSCAB/PhoU complex, which transduces phosphate transporter conformational changes to PhoR. However, in B. subtilis, an alternative mechanism links PhoR regulation to wall teichoic acid metabolism, where biosynthetic intermediates modulate PhoR autokinase activity.
PhoS regulates biofilm formation in Bacillus sp and is induced under phosphate limitation [220].
Disruption of phoS reduced extracellular polymeric substance (EPS) production, whereas overexpression increased EPS levels, confirming its role in biofilm synthesis. PhoS enhances translation of PhoP mRNA by preventing an inhibitory structure at its 5’UTR, forming a self-reinforcing autoregulatory loop that allows Bacillus sp to adapt to phosphate scarcity. Through PhoP, PhoS promotes biofilm formation by influencing the expression of operons involved in cell wall metabolism, such as tuaA-H, which facilitates teichuronic acid synthesis. Additionally, PhoS potentially affects biofilm development through the PhoP-regulated comQ-X-P-A operon, impacting quorum sensing and surfactin signaling. While the deletion of phoS weakly reduces biofilm formation, its overexpression strongly enhances it, likely due to the regulatory loop amplifying PhoP levels. Transcriptomic analyses reveal PhoS modulates key biofilm-related genes, though it does not directly target eps or tapA-sipW-tasA operons in Bacillus subtilis and Bacillus velezensis strain FZB42.
4.4.3. SrbA
The small RNA SrbA plays a crucial role in P. aeruginosa biofilm formation. A srbA deletion mutant exhibited a 66% reduction in biofilm formation while maintaining normal surface attachment in C. elegans slow-killing model. Complementation of srbA restored wild-type biofilm levels, indicating SrbA’s essential function. Genome-wide analysis (using TargetRNA2 and BLASTn) identified 61 putative mRNA targets of SrbA, including genes linked to biofilm formation, motility, metabolism, and stress response, suggesting broad post-transcriptional regulation. RT-qPCR revealed significant transcriptomic changes in the srbA mutant during biofilm growth, with 12 genes downregulated ≥4-fold and 7 upregulated ≥2-fold. These findings suggest SrbA modulates biofilm formation by influencing mRNA stability and translation efficiency, potentially via interactions with the ribosome binding site or RNase E recruitment [221].
5. Role of Small RNA in Quorum Sensing
Bacteria regulate population density through a process known as quorum sensing (QS), which involves the production, release, accumulation, and detection of extracellular signaling molecules termed autoinducers (AIs) [14, 16, 222]. Quorum sensing enables bacterial communities to coordinate responses to chemical signals, promoting cooperative behaviors that enhance fitness [16, 223]. These include coordinated virulence, evadiing host immune responses, and establishing infections [224]. This mechanism is present in both Gram-positive and Gram-negative pathogens, though the signaling molecules triggering QS circuits differ between them. Gram positive bacteria typically respond to processed oligopeptides [53] whereas Gram-negative bacteria respond to homoserine lactones as autoinducers [16]. Previous works on Pseudomonas aeruginosa [225], Staphylococcus aureus [226], and Vibrio cholerae [227], reveals that these pathogenic use QS to regulate the production of virulence factors, including toxins, proteases, siderophores, and biofilm formation, which enhance their survival within the host . QS also facilitates immune evasion by regulating the expression of immune-modulatory molecules, allowing bacteria to remain undetected until they reach a critical population density. Additionally, QS-mediated biofilm formation protects bacteria from antibiotics and immune clearance, contributing to chronic infections [209, 228]. Emerging evidence suggests that host organisms can interfere with QS through quorum quenching, a strategy in which the host degrades or inhibits bacterial signaling molecules to prevent infection. Understanding QS in host-pathogen interactions is essential for developing novel anti-virulence therapies that target bacterial communication without promoting antibiotic resistance.
This section will explore the mechanism of quorum sensing and its role in bacterial pathogenesis and host-pathogen interactions, focusing on two Gram-negative representatives, Vibrio sp. and Pseudomonas aeruginosa, and one Gram-positive representative, Staphylococcus aureus.
5.1. Qrr 1-5
Quorum sensing (QS) in Vibrio harveyi and Vibrio cholerae is controlled by small regulatory RNAs (sRNAs) called quorum regulatory RNAs (Qrrs) and the RNA chaperone Hfq. This system enables bacteria to synchronize group behaviors, including bioluminescence, virulence factors production, type VI secretion, and biofilm formation as a function of cell density. V. harveyi produces three autoinducers—an acyl homoserine lactone-HAI-1 (also known as harveyi auto inducer 1), a furanosyl borate diester-AI-2 (also known as autoinducer-2) , and (S)-3-hydroxytridecan-4-one- CAI-1 (also known as cholerae autoinducer 1)—synthesized by LuxM, LuxS, and CqsA, respectively [229-231]. These autoinducers are detected by membrane-bound two-component sensors LuxN, LuxQ, and CqsS (Figure 4). The autoinducers regulate QS through a phosphorelay system. At low cell density, when autoinducer concentrations are minimal, these sensors function as kinases, phosphorylating LuxU, which subsequently phosphorylates LuxO. Phosphorylated LuxO (LuxO–P), in conjunction with σ⁵⁴, activates the transcription of five Qrr sRNAs (Qrr1–5) [230]. These Qrr sRNAs bind to Hfq and interact with target mRNAs through specific base-pairing to regulate gene expression via catalytic degradation, coupled degradation, sequestration, or activation-induced degradation. Each Qrr sRNA contains four stem-loops, with the first and second being critical for base pairing and stability. This regulatory network represses the master QS regulator LuxR, preventing QS-induced gene expression at low cell. At high cell density, accumulated autoinducers switch the sensors from kinases to phosphatases, resulting in LuxO dephosphorylation and subsequent cessation of Qrr transcription. This permits LuxR translation and the activation of QS-regulated genes, leading to behaviors such as bioluminescence and biofilm formation (Figure 4).
In V. cholerae, the QS system operates similarly but lacks the HAI-1 pathway, relying on AI-2 and CAI-1 autoinducers [232, 233]. The phosphorelay system involving LuxU and LuxO regulates QS gene expression. However, unlike V. harveyi, V. cholerae produces four Qrr sRNAs (Qrr1–4), which redundantly repress HapR, the homolog of LuxR. Additionally, the VarS–VarA two-component sensory system modulates QS by regulating the activity of the CsrA protein, which indirectly influences LuxO activity [234]. At low cell density, CsrA enhances LuxO–P activity, promoting Qrr-mediated repression of HapR. At high cell density, VarS–VarA inhibits CsrA, allowing HapR expression, which suppresses virulence factor production and biofilm formation.
A key player of Vibrio cholerae virulence is the type VI secretion system (T6SS). T6SS is a QS-regulated, contact-dependent protein delivery apparatus that can eliminate competing cells [235, 236]. Structurally, T6SS assembles into a membrane-spanning, spear-like device loaded with toxic effector proteins [237]. Upon deployment, the effectors penetrate competitor cell walls. To prevent self-toxicity, T6SS-active cells produce immunity proteins that neutralize the toxic effectors [238]. Additional protective mechanisms, such as exopolysaccharide and capsular polysaccharide production, also safeguard against incoming T6SS-mediated attacks [239]. The Qrr sRNAs repress t6ss expression via two mechanisms: direct repression of the primary t6ss gene cluster by interacting with their mRNA, and indirect repression by inhibiting HapR, the HCD activator of auxiliary t6ss gene clusters [235]. Since T6SS toxin deployment requires direct cell-to-cell contact, limiting maximal T6SS machinery production to HCD may enhance killing efficiency and reduce energy expenditure. Interestingly, Huber et al. identified an RNA sponge, QrrX, which base-pairs with and inactivates the Qrr1-4 sRNAs [240]. The transcription of qrrX is driven by QrrT, a previously uncharacterized LysR-type transcriptional regulator. Findings from this study suggest that QrrX and QrrT play essential roles in facilitating the transition from individual to community behaviors in Vibrio cholerae.
5.2. PhrS and ReaL
Pseudomonas aeruginosa is an opportunistic pathogen that causes severe infections in immunocompromised individuals and cystic fibrosis patients [241]. . In Pseudomonas aeruginosa, four interconnected QS systems—las, rhl, pqs, and iqs—regulate over 300 virulence-related genes [225, 242]. The las and rhl systems use N-acyl homoserine lactone (AHL) signals, with LasI producing 3-oxo-C12-HSL to activate LasR, leading to the expression of virulence factors like elastases and exotoxins. The rhl system, with RhlI producing C4-HSL, interacts with RhlR to regulate factors such as rhamnolipid and hydrogen cyanide. The pqs system synthesizes quinolones (PQS signals) via PqsABCD, activating PqsR. (PQS) acts as a critical link between the Las and Rhl systems, regulating genes involved in biofilm formation, iron acquisition, and production of the redox-active toxin pyocyanin (PYO) [243, 244]. PYO contributes to pathogenicity by generating reactive oxygen species (ROS), inducing oxidative stress, and damaging host tissues, while also exhibiting antimicrobial activity against competing microorganisms [245]The iqs system, with AmbBCDE, produces IQS signals. These QS systems function hierarchically, with las at the top, regulating rhl and pqs, while pqs also modulates las and rhl.
The small regulatory RNA (sRNA) PhrS plays a central role in modulating QS and virulence in P. aeruginosa [246]. Its expression is controlled by the oxygen-responsive transcription factor ANR and is enhanced during hypoxia or stationary phase. A putative ANR binding site upstream of the phrS promoter has been identified, and phrS expression is abolished in ANR-deficient mutants [246]. Additionally, Hfq indirectly regulates phrS by stabilizing ANR. PhrS promotes the translation of pqsR—a key activator of PQS synthesis—through structural rearrangements in hypoxic conditions, ultimately enhancing PQS and PYO production. PYO contributes to immune evasion through ROS generation and modulation of IL-8-driven immune responses. PhrS-deficient strains exhibit impaired PYO production, highlighting its critical role in P. aeruginosa virulence. In addition, PhrS downregulates the expression of cytochrome bo3 oxidase genes (cyoAB), facilitating a shift in oxygen utilization pathways under low oxygen [246]. By overcoming oxygen-dependent suppression of PQS, PhrS supports QS-dependent gene expression under hypoxia, a condition common in biofilms and cystic fibrosis lung infections. Furthermore, PhrS-mediated PQS synthesis promotes extracellular DNA release, a crucial component for biofilm stability. These findings suggest that targeting PhrS may offer a novel strategy to mitigate P. aeruginosa infections.
In addition to PhrS, the sRNA ReaL in Pseudomonas aeruginosa plays a role in virulence by linking the las and pqs quorum sensing (QS) systems [247]. Deletion of reaL reduces virulence in the Galleria mellonella infection model, while its overexpression leads to a hyper-virulent phenotype. ReaL is negatively regulated by the las QS regulator LasR and enhances the synthesis of the pqs quinolone signal PQS by post-transcriptionally activating pqsC. The region from −13 to +32 of pqsC mRNA is predicted to form a stable stem-loop structure, sequestering the Shine-Dalgarno (SD) sequence and the AUG start codon through base pairing with the RII region, effectively acting as an anti-SD/AUG element and repressing pqsC translation. When ReaL binds to the RI-RII region of pqsC, it induces a conformational change that extrudes a loop containing the SD and AUG sequences. This structural rearrangement facilitates the recruitment of the 30S ribosomal subunit to the mRNA, enabling translation initiation [247]. This affects key virulence traits such as pyocyanin production, biofilm formation, and swarming motility. Additionally, ReaL responds to host-relevant conditions like temperature and oxygen levels and is upregulated in the stationary phase by the alternative σ factor RpoS. These regulatory mechanisms fine-tune PQS synthesis and contribute to P. aeruginosa virulence.
5.3. AmiL and PqsS
In addition to PhrS and ReaL discussed above, recent studies have identified AmiL, a QS-regulatory sRNA that modulates virulence in Pseudomonas aeruginosa by targeting genes involved in key virulence factors and phenotypes such as pyocyanin, elastase, rhamnolipid, biofilm formation, swarming motility, hemolysin, and cytotoxicity [248]. However, unlike AmiL and PqsS, which positively regulate pyocyanin, AmiL acts as a negative regulator. The 100 nt sRNA AmiL is a leader of the amiEBCRS operon involved in biofilm formation [249]. Using transcriptomics, Pu et al. revealed that AmiL expression is negatively regulated by RhlR [248]. As discussed above, PhrS directly targets the PhzC (a member of the operon responsible for synthesis of Pyocyanin) and regulates lasI and rhlI expression [246, 250]. This defines a new las/rhl (RhlR)-AmiL-PhzC/las/rhl signaling cascade, emphasizing AmiL’s central role in PAO1 virulence. While AmiL downregulates lasI during early growth, it upregulates rhlI and C4-HSL production in later stages [248]. It also promotes swarming motility by enhancing rhamnolipid production. This dual role in las and rhl regulation highlights AmiL’s function in fine-tuning P. aeruginosa’s QS-sRNA network to balance virulence.
Another Hfq dependent small RNA PqsS was recently discovered in P. aeruginosa PAO1, which enhances bacterial pathogenicity by regulating the Pseudomonas quinolone signal (PQS) quorum sensing (pqs QS) system [251]. PqsS promotes acute infections by inducing host cell death and stimulating rhamnolipid-mediated swarming motility, while simultaneously reducing chronic infection traits such as biofilm formation and antibiotic resistance. Mechanistically, PqsS represses pqsL transcript, leading to increased PQS levels and reinforcing pqs QS [251]. Additionally, a PQS-rich environment upregulates PqsS expression, establishing a positive feedback loop. Further analysis revealed that PqsS interacts with pqsL mRNA, recruiting RNase E to facilitate its degradation. These findings provide valuable insights into P. aeruginosa pathogenesis and offer potential avenues for targeted therapeutic strategies
5.4. SprD
The Gram-positive pathogen Staphylococcus aureus utilize quorum sensing (QS) to regulate virulence and biofilm formation, primarily through the Agr (Accessory gene regulator) system. The agr system operates via a two-component signaling mechanism involving the autoinducing peptide (AIP) and the AgrC-AgrA regulatory circuit. AIP is synthesized as a precursor and processed into its active form, which binds to the membrane-bound sensor kinase AgrC, triggering a phosphorylation cascade that activates the response regulator AgrA. This activation leads to the expression of RNAIII, a regulatory RNA that controls virulence gene expression, including toxins and exoenzymes, while repressing surface proteins associated with early biofilm formation [95].
In addition to RNAIII, which was discussed in section 2.2.2, another small RNA involved in quorum sensing in Staphylococcus is the small pathogenicity island RNA D (SprD) [252]. SprD is crucial for the virulence of Staphylococcus aureus, as its deletion in a murine sepsis model resulted in 100% survival by day 21, compared to complete mortality in mice infected with the wild-type strain. Complementing the mutant with SprD partially restored virulence (50% survival). Moreover, mice infected with the ΔsprD strain had smaller, uniformly discolored kidneys, whereas wild-type infections caused swollen, abscess-filled kidneys [252]. Bacterial counts revealed reduced bacterial persistence in the absence of SprD. While SprD regulates the immune-evasion protein Sbi, altering Sbi expression alone did not affect virulence, suggesting SprD targets additional factors essential for pathogenesis. The regulation of sbi mRNA by SprD in Staphylococcus aureus involves direct antisense pairing, as shown by in silico predictions and confirmed through gel retardation assays. SprD binds a specific region of sbi mRNA near its Shine-Dalgarno (SD) sequence and AUG initiation codon, forming a stable duplex that prevents ribosome loading and translation initiation [252]. Deletion mutants of both SprD and sbi mRNA abolished this interaction, confirming the necessity of these binding regions.
Taken together Small regulatory RNAs are key regulators of quorum sensing in pathogens like Escherichia coli, Vibrio spp., Pseudomonas aeruginosa, and Staphylococcus aureus. By modulating quorum sensing components, sRNAs influence bacterial communication, virulence, and biofilm formation, aiding adaptation and immune evasion. Their role in host-pathogen interactions makes them promising targets for antimicrobial strategies to disrupt quorum sensing-mediated virulence.
6. Discussion
The study of bacterial small regulatory RNAs has greatly advanced due to the development of high-throughput sequencing technologies, allowing for a more comprehensive understanding of sRNA networks and their regulatory roles. In Escherichia coli, RNA sequencing (RNA-seq) has identified approximately 108 sRNAs, while sRNA interactome profiling has revealed between 1,600 and 1,900 sRNA-mRNA interactions. These extensive regulatory networks parallel the complexity of transcription factor interactions, which encompass 217 transcription factors forming 3,466 chromosomal interactions. Many transcriptional regulatory circuits integrate sRNAs as key components, enabling an additional layer of post-transcriptional control. Similar to transcription factors, sRNAs often function as regulatory "hubs," influencing a broad array of target RNAs and coordinating biologically cohesive groups of mRNAs that collectively shape phenotypic outcomes. The development of sophisticated sequencing-based methodologies such as RIL-seq (RNA Interaction by Ligation and Sequencing) [253], GRIL-seq (Global RNA Interaction by Ligation and Sequencing) [254], Hi-GRIL-seq (High-Throughput GRIL-seq) [183], and [255] has revolutionized our ability to map these complex interactions. These techniques provide valuable insights into the intricate RNA regulatory networks that govern bacterial stress responses, antibiotic resistance, and virulence. RIL-seq, for instance, captures transcriptome-wide RNA-RNA interactions mediated by RNA-binding proteins (RBPs) such as Hfq, revealing complex sRNA-mRNA interaction networks. GRIL-seq, on the other hand, specifically focuses on identifying sRNA interactions without relying on RBP immunoprecipitation, using in vivo proximity ligation to directly capture RNA duplexes. Meanwhile, Hfq-RIP-seq enables genome-wide identification of Hfq-associated RNAs, further illuminating the role of this global RNA chaperone in post-transcriptional regulation. Together, these methodologies have significantly expanded our understanding of bacterial sRNA networks and their regulatory influence over critical cellular processes.
Recent advances in bacterial stress response and pathogenesis research have unveiled sophisticated regulatory networks that enable bacteria to survive harsh conditions, including extreme temperatures, oxidative stress, nutrient deprivation, and antibiotic exposure [256, 257]. A key breakthrough has been the discovery of novel small RNAs (sRNAs) that fine-tune gene expression in response to environmental fluctuations, allowing rapid adaptation. Additionally, the role of phase separation in stress granule formation has emerged as a critical mechanism for bacterial survival under stress [258]. Advances in multi-omics approaches, such as single-cell RNA sequencing and proteomics, have provided deeper insights into stress-induced regulatory circuits. Furthermore, CRISPR-based tools have been employed to dissect stress-related genetic pathways, revealing new targets for antimicrobial development [259]. These findings not only expand our understanding of bacterial resilience but also offer potential strategies for combating antibiotic resistance and improving industrial microbial applications.
Similarly, emerging trends in quorum sensing (QS) research have unveiled novel strategies to modulate bacterial communication, offering potential therapeutic avenues against pathogenicity [14]. Additionally, the integration of quorum sensing inhibitors (QSIs) with traditional antibiotics has shown synergistic effects, enhanced antibacterial efficacy and providing new strategies for clinical treatment of bacterial infections [260]. Furthermore, the application of exogenous QS signaling molecules, such as acyl-homoserine lactones (AHLs), has been explored to enhance electrochemical activities in bioelectrochemical systems, indicating QS's potential in bioengineering applications [261].Collectively, these advancements underscore the pivotal role of QS in bacterial behavior and its promising exploitation in medical and industrial contexts.
Ongoing advancements highlight the crucial role of sRNAs in bacterial adaptation to antibiotics by regulating resistance and tolerance mechanisms. Exposure to sub-minimal inhibitory concentrations (sub-MICs) of antibiotics induces broad transcriptional and proteomic changes, including alterations in sRNA expression profiles [262]. This adaptive response enables bacteria to evade growth inhibition and develop antibiotic tolerance. Specific sRNAs have been implicated in antibiotic resistance across multiple species. In Salmonella Typhimurium, for example, tigecycline treatment induces the sRNA SroA [263], whose deletion increases susceptibility to the drug, while ectopic expression restores resistance. Similarly, in Clostridia, exposure to clindamycin induces an sRNA located upstream of an ABC transporter gene, conferring resistance in Staphylococcus species [264]. Transcriptomic analyses of antibiotic-resistant pathogens, including Mycobacterium tuberculosis, Methicillin-resistant Staphylococcus aureus (MRSA), and Pseudomonas putida, have demonstrated that different antibiotics elicit unique sRNA expression profiles [265-267]. Some antibiotics induce extensive sRNA regulatory changes, while others influence only a limited subset of sRNAs. In E. coli, antibiotic stress triggers the activation of the alternative sigma factor RpoS through Hfq-dependent sRNAs such as RprA, which governs a range of stress response genes [268]. RpoS activation facilitates mutagenesis by upregulating error-prone DNA polymerase IV [269] and downregulating the DNA repair protein MutS via the sRNA SdsR. These processes enhance genetic diversity, accelerating the evolution of antibiotic resistance. Furthermore, RpoS and its associated sRNAs contribute to horizontal gene transfer by promoting plasmid conjugation, a key mechanism in the dissemination of resistance genes among bacterial populations [270]. The interplay between antibiotic exposure, sRNA regulation, and genetic adaptation underscores the critical role of sRNAs in bacterial survival and evolutionary dynamics in response to antimicrobial pressure.
Expanding our understanding of sRNA dynamics reveals new strategies to combat antimicrobial resistance by targeting resistance-conferring sRNAs or disrupting key regulatory networks. [271]. Synthetic antisense RNAs, for instance, could be designed to selectively inhibit sRNAs that enhance bacterial survival under antibiotic stress. Additionally, exploiting sRNA interactions could sensitize bacteria to existing antibiotics by disrupting tolerance mechanisms, thereby improving treatment efficacy. Advances in computational tools such as sRNAdeep [272] and TargetRNA3 [273] have facilitated the accurate prediction and characterization of sRNAs, enabling more precise functional studies. These machine learning-based approaches have significantly improved the identification of bacterial sRNAs and their regulatory targets, providing researchers with powerful tools for elucidating RNA-based regulatory mechanisms. The continued development of high-throughput RNA sequencing techniques and computational models will be instrumental in advancing our understanding of sRNA-mediated gene regulation and its implications for bacterial pathogenesis and antibiotic resistance. As antimicrobial resistance (AMR) continues to pose a significant global health challenge, further research into sRNA-mediated regulatory mechanisms will be essential for developing novel therapeutic strategies aimed at mitigating the impact of drug-resistant bacterial infections.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, writing—original draft preparation. writing—review and editing, visualization, supervision-Rajdeep Banerjee.
Funding
The author has not received any funding for writing this review.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Raghavan, S.; Kim, K.-S. Host immunomodulation strategies to combat pandemic-associated antimicrobial-resistant secondary bacterial infections. Int. J. Antimicrob. Agents 2024, 64, 107308. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Gallo, R.L. Innate immune defense system of the skin. Veter- Dermatol. 2013, 24, 32–e9. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Feng, W.; Wang, J.; Wang, R.; Zhang, B.; Bo, L.; Chen, Z.-S.; Yang, H.; Sun, L. Antimicrobial peptides for combating drug-resistant bacterial infections. Drug Resist. Updat. 2023, 68, 100954. [Google Scholar] [CrossRef]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The Roles of Antimicrobial Peptides in Innate Host Defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [PubMed]
- Dlozi, P.N.; Gladchuk, A.; Crutchley, R.D.; Keuler, N.; Coetzee, R.; Dube, A. Cathelicidins and defensins antimicrobial host defense peptides in the treatment of TB and HIV: Pharmacogenomic and nanomedicine approaches towards improved therapeutic outcomes. Biomed. Pharmacother. 2022, 151, 113189–113189. [Google Scholar] [CrossRef]
- A Lewis, L.; Ram, S. Meningococcal disease and the complement system. Virulence 2013, 5, 98–126. [Google Scholar] [CrossRef]
- Subashchandrabose, S.; Mobley, H.L.T. Virulence and Fitness Determinants of Uropathogenic Escherichia coli. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Murdoch, C.C.; Skaar, E.P. Nutritional immunity: the battle for nutrient metals at the host–pathogen interface. Nat. Rev. Microbiol. 2022, 20, 657–670. [Google Scholar] [CrossRef]
- Bengoechea, J.A.; Sa Pessoa, J. Klebsiella pneumoniae infection biology: living to counteract host defences. FEMS Microbiology Reviews 2018, 43, 123–144. [Google Scholar] [CrossRef]
- Cooper, A.M. Cell-Mediated Immune Responses in Tuberculosis. Annu. Rev. Immunol. 2009, 27, 393–422. [Google Scholar] [CrossRef]
- Hornef, M.W.; Wick, M.J.; Rhen, M.; Normark, S. Bacterial strategies for overcoming host innate and adaptive immune responses. Nat. Immunol. 2002, 3, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; McFadden, G. Anti-Immunology: Evasion of the Host Immune System by Bacterial and Viral Pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef]
- Rutherford, S.T.; Bassler, B.L. Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Whiteley, M.; Diggle, S.P.; Greenberg, E.P. Progress in and promise of bacterial quorum sensing research. Nature 2017, 551, 313–320. [Google Scholar] [CrossRef]
- Uddin, T.M.; Chakraborty, A.J.; Khusro, A.; Zidan, B.R.M.; Mitra, S.; Bin Emran, T.; Dhama, K.; Ripon, K.H.; Gajdács, M.; Sahibzada, M.U.K.; et al. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J. Infect. Public Heal. 2021, 14, 1750–1766. [Google Scholar] [CrossRef]
- Sun, J.; Deng, Z.; Yan, A. Bacterial multidrug efflux pumps: Mechanisms, physiology and pharmacological exploitations. Biochemical and Biophysical Research Communications 2014, 453, 254–267. [Google Scholar] [CrossRef]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol Spectr 2016, 4. [Google Scholar] [CrossRef]
- Raina, M., et al., Dual-Function RNAs. Microbiol Spectr, 2018. 6(5).
- Gottesman, S.; Storz, G. Bacterial Small RNA Regulators: Versatile Roles and Rapidly Evolving Variations. Cold Spring Harb. Perspect. Biol. 2010, 3, a003798. [Google Scholar] [CrossRef]
- Storz, G.; Vogel, J.; Wassarman, K.M. Regulation by Small RNAs in Bacteria: Expanding Frontiers. Mol. Cell 2011, 43, 880–891. [Google Scholar] [CrossRef]
- Svenningsen, S.L. Small RNA-Based Regulation of Bacterial Quorum Sensing and Biofilm Formation. Microbiol. Spectr. 2018, 6. [Google Scholar] [CrossRef]
- Lenz, D.H., et al., The Small RNA Chaperone Hfq and Multiple Small RNAs Control Quorum Sensing in Vibrio harveyi and Vibrio cholerae. Cell, 2004. 118(1): p. 69-82.
- Sy, B.M.; Tree, J.J. Small RNA Regulation of Virulence in Pathogenic Escherichia coli. Front. Cell. Infect. Microbiol. 2021, 10. [Google Scholar] [CrossRef]
- Li, W.; Ying, X.; Lu, Q.; Chen, L. Predicting sRNAs and Their Targets in Bacteria. Genom. Proteom. Bioinform. 2012, 10, 276–284. [Google Scholar] [CrossRef]
- Waters, L.S.; Storz, G. Regulatory RNAs in Bacteria. Cell 2009, 136, 615–628. [Google Scholar] [CrossRef]
- Grešová, K.; Alexiou, P.; Giassa, I.-C. Small RNA Targets: Advances in Prediction Tools and High-Throughput Profiling. Biology 2022, 11, 1798. [Google Scholar] [CrossRef]
- Fang, F.C.; Frawley, E.R.; Tapscott, T.; Vázquez-Torres, A. Bacterial Stress Responses during Host Infection. Cell Host Microbe 2016, 20, 133–143. [Google Scholar] [CrossRef]
- Everest, P. Stress and bacteria: microbial endocrinology. Gut 2007, 56, 1037–1038. [Google Scholar] [CrossRef]
- Gottesman, S. Trouble is coming: Signaling pathways that regulate general stress responses in bacteria. J. Biol. Chem. 2019, 294, 11685–11700. [Google Scholar] [CrossRef]
- Sanyal, S., et al., Polyphosphate kinase 1, a central node in the stress response network of Mycobacterium tuberculosis, connects the two-component systems MprAB and SenX3-RegX3 and the extracytoplasmic function sigma factor, sigma E. Microbiology (Reading), 2013. 159(Pt 10): p. 2074-2086.
- Sharma, A.K.; Basu, J.; Kundu, M.; Mandal, S.; Mukhopadhyay, J.; Gupta, S.; Banerjee, R.; Chatterjee, A. MtrA, an essential response regulator of the MtrAB two-component system, regulates the transcription of resuscitation-promoting factor B of Mycobacterium tuberculosis. Microbiology 2015, 161, 1271–1281. [Google Scholar] [CrossRef]
- Durand, S.; Storz, G. Reprogramming of anaerobic metabolism by the FnrS small RNA. Mol. Microbiol. 2010, 75, 1215–1231. [Google Scholar] [CrossRef]
- Papenfort, K.; Storz, G. Insights into bacterial metabolism from small RNAs. Cell Chem. Biol. 2024, 31, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Nirban, R.; Dutta, T. MTS1338 in Mycobacterium tuberculosis promotes detoxification of reactive oxygen species under oxidative stress. Tuberculosis 2021, 131, 102142. [Google Scholar] [CrossRef]
- Villa, J.K.; Han, R.; Tsai, C.-H.; Chen, A.; Sweet, P.; Franco, G.; Vaezian, R.; Tkavc, R.; Daly, M.J.; Contreras, L.M. A small RNA regulates pprM, a modulator of pleiotropic proteins promoting DNA repair, in Deinococcus radiodurans under ionizing radiation. Sci. Rep. 2021, 11, 12949. [Google Scholar] [CrossRef]
- Chambers, J.R.; Sauer, K. Small RNAs and their role in biofilm formation. Trends Microbiol. 2013, 21, 39–49. [Google Scholar] [CrossRef]
- Bak, G.; Lee, J.; Suk, S.; Kim, D.; Lee, J.Y.; Kim, K.-S.; Choi, B.-S.; Lee, Y. Identification of novel sRNAs involved in biofilm formation, motility and fimbriae formation in Escherichia coli. Sci. Rep. 2015, 5, srep15287. [Google Scholar] [CrossRef]
- Sy, B.M.; Lan, R.; Tree, J.J. Early termination of the Shiga toxin transcript generates a regulatory small RNA. Proc. Natl. Acad. Sci. 2020, 117, 25055–25065. [Google Scholar] [CrossRef]
- Djapgne, L.; Oglesby, A.G. Impacts of Small RNAs and Their Chaperones on Bacterial Pathogenicity. Front. Cell. Infect. Microbiol. 2021, 11, 604511. [Google Scholar] [CrossRef]
- Danger, J.L.; Makthal, N.; Kumaraswami, M.; Sumby, P. The FasX Small Regulatory RNA Negatively Regulates the Expression of Two Fibronectin-Binding Proteins in Group A Streptococcus. J. Bacteriol. 2015, 197, 3720–3730. [Google Scholar] [CrossRef]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- (WHO), W.H.O., WHO Bacterial Priority Pathogens List, 2024. 2024.
- Lopatkin, A.J.; Sysoeva, T.A.; You, L. Dissecting the effects of antibiotics on horizontal gene transfer: Analysis suggests a critical role of selection dynamics. BioEssays 2016, 38, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, C.; Grohmann, E. Horizontal Gene Transfer of Antibiotic Resistance Genes in Biofilms. Antibiotics 2023, 12, 328. [Google Scholar] [CrossRef]
- Mediati, D.G.; Wu, S.; Wu, W.; Tree, J.J. Networks of Resistance: Small RNA Control of Antibiotic Resistance. Trends Genet. 2020, 37, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dersch, P.; Khan, M.A.; Mühlen, S.; Görke, B. Roles of Regulatory RNAs for Antibiotic Resistance in Bacteria and Their Potential Value as Novel Drug Targets. Front. Microbiol. 2017, 8, 803. [Google Scholar] [CrossRef]
- Bejerano-Sagie, M.; Xavier, K.B. The role of small RNAs in quorum sensing. Curr. Opin. Microbiol. 2007, 10, 189–198. [Google Scholar] [CrossRef]
- Liu, X., P. Zhou, and R. Wang, Small RNA-mediated switch-like regulation in bacterial quorum sensing. IET Syst Biol, 2013. 7(5): p. 182-7.
- Fu, H.; Elena, R.C.; Marquez, P.H. The roles of small RNAs: insights from bacterial quorum sensing. ExRNA 2019, 1, 1–8. [Google Scholar] [CrossRef]
- Federle, M.J. and B.L. Bassler, Interspecies communication in bacteria. J Clin Invest, 2003. 112(9): p. 1291-9.
- Arnold, B.J.; Huang, I.-T.; Hanage, W.P. Horizontal gene transfer and adaptive evolution in bacteria. Nat. Rev. Microbiol. 2021, 20, 206–218. [Google Scholar] [CrossRef]
- Updegrove, T.B., S.A. Shabalina, and G. Storz, How do base-pairing small RNAs evolve? FEMS Microbiol Rev, 2015. 39(3): p. 379-91.
- Raad, N.; Tandon, D.; Hapfelmeier, S.; Polacek, N. The stationary phase-specific sRNA FimR2 is a multifunctional regulator of bacterial motility, biofilm formation and virulence. Nucleic Acids Res. 2022, 50, 11858–11875. [Google Scholar] [CrossRef]
- Meng, X., et al., Functions of Small Non-Coding RNAs in Salmonella-Host Interactions. Biology (Basel), 2022. 11(9).
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III Secretion Systems and Disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef]
- Cakar, F.; Golubeva, Y.A.; Vanderpool, C.K.; Slauch, J.M. The Small RNA MicC Downregulates hilD Translation To Control the Salmonella Pathogenicity Island 1 Type III Secretion System in Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2022, 204, e0037821. [Google Scholar] [CrossRef] [PubMed]
- Abdulla, S.Z.; Kim, K.; Azam, M.S.; Golubeva, Y.A.; Cakar, F.; Slauch, J.M.; Vanderpool, C.K. Small RNAs Activate Salmonella Pathogenicity Island 1 by Modulating mRNA Stability through the hilD mRNA 3′ Untranslated Region. J. Bacteriol. 2023, 205. [Google Scholar] [CrossRef] [PubMed]
- Ostrik, A.A.; Azhikina, T.L.; Salina, E.G. Small Noncoding RNAs and Their Role in the Pathogenesis of Mycobacterium tuberculosis Infection. Biochem. (Moscow) 2021, 86, S109–S119. [Google Scholar] [CrossRef]
- Peñaloza, D.; Acuña, L.G.; Barros, M.J.; Núñez, P.; Montt, F.; Gil, F.; Fuentes, J.A.; Calderón, I.L. The Small RNA RyhB Homologs from Salmonella Typhimurium Restrain the Intracellular Growth and Modulate the SPI-1 Gene Expression within RAW264.7 Macrophages. Microorganisms 2021, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Melican, K.; Sandoval, R.M.; Kader, A.; Josefsson, L.; Tanner, G.A.; Molitoris, B.A.; Richter-Dahlfors, A. Uropathogenic Escherichia coli P and Type 1 Fimbriae Act in Synergy in a Living Host to Facilitate Renal Colonization Leading to Nephron Obstruction. PLOS Pathog. 2011, 7, e1001298. [Google Scholar] [CrossRef]
- Snyder, J.A., et al., Coordinate Expression of Fimbriae in Uropathogenic Escherichia coli. Infection and Immunity, 2005. 73(11): p. 7588-7596.
- Müller, C.M.; Åberg, A.; Straseviçiene, J.; Emődy, L.; Uhlin, B.E.; Balsalobre, C. Type 1 Fimbriae, a Colonization Factor of Uropathogenic Escherichia coli, Are Controlled by the Metabolic Sensor CRP-cAMP. PLOS Pathog. 2009, 5, e1000303. [Google Scholar] [CrossRef]
- Miyazaki, J.; Ba-Thein, W.; Kumao, T.; Yasuoka, M.O.; Akaza, H.; Hayshi, H. Type 1, P and S fimbriae, and afimbrial adhesin I are not essential for uropathogenic Escherichia coli to adhere to and invade bladder epithelial cells. FEMS Immunol. Med Microbiol. 2002, 33, 23–26. [Google Scholar] [CrossRef]
- Bryan, A., et al., Regulation of type 1 fimbriae by unlinked FimB- and FimE-like recombinases in uropathogenic Escherichia coli strain CFT073. Infect Immun, 2006. 74(2): p. 1072-83.
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Wiles, T.J.; Kulesus, R.R.; Mulvey, M.A. Origins and virulence mechanisms of uropathogenic Escherichia coli. Exp. Mol. Pathol. 2008, 85, 11–19. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Z.; Zheng, L.; Gong, Z.; Li, Y.; Jin, Y.; Huang, Y.; Chi, M. Urinary Tract Infections Caused by Uropathogenic Escherichia coli: Mechanisms of Infection and Treatment Options. Int. J. Mol. Sci. 2023, 24, 10537. [Google Scholar] [CrossRef]
- Wright, K.J.; Hultgren, S.J. Sticky Fibers and Uropathogenesis: Bacterial Adhesins in the Urinary Tract. Futur. Microbiol. 2006, 1, 75–87. [Google Scholar] [CrossRef]
- Biggel, M., et al., Horizontally acquired papGII-containing pathogenicity islands underlie the emergence of invasive uropathogenic Escherichia coli lineages. Nat Commun, 2020. 11(1): p. 5968.
- Bien, J.; Sokolova, O.; Bozko, P. Role of UropathogenicEscherichia coliVirulence Factors in Development of Urinary Tract Infection and Kidney Damage. Int. J. Nephrol. 2012, 2012, 1–15. [Google Scholar] [CrossRef]
- Khandige, S., et al., sRNA-Mediated Regulation of P-Fimbriae Phase Variation in Uropathogenic Escherichia coli. PLoS Pathog, 2015. 11(8): p. e1005109.
- Holden, N.J.; Gally, D.L. Switches, cross-talk and memory in Escherichia coli adherence. J. Med Microbiol. 2004, 53, 585–593. [Google Scholar] [CrossRef]
- Bessaiah, H.; Pokharel, P.; Loucif, H.; Kulbay, M.; Sasseville, C.; Habouria, H.; Houle, S.; Bernier, J.; Massé, É.; Van Grevenynghe, J.; et al. The RyfA small RNA regulates oxidative and osmotic stress responses and virulence in uropathogenic Escherichia coli. PLOS Pathog. 2021, 17, e1009617. [Google Scholar] [CrossRef]
- Fris, M.E.; Broach, W.H.; Klim, S.E.; Coschigano, P.W.; Carroll, R.K.; Caswell, C.C.; Murphy, E.R. Sibling sRNA RyfA1 Influences Shigella dysenteriae Pathogenesis. Genes 2017, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Sievers, S., et al., A multicopy sRNA of Listeria monocytogenes regulates expression of the virulence adhesin LapB. Nucleic Acids Res, 2014. 42(14): p. 9383-98.
- Sievers, S.; Lund, A.; Menendez-Gil, P.; Nielsen, A.; Mollerup, M.S.; Nielsen, S.L.; Larsson, P.B.; Borch-Jensen, J.; Johansson, J.; Kallipolitis, B.H. The multicopy sRNA LhrC controls expression of the oligopeptide-binding protein OppA inListeria monocytogenes. RNA Biol. 2015, 12, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Meng, X., et al., Small non-coding RNA STnc640 regulates expression of fimA fimbrial gene and virulence of Salmonella enterica serovar Enteritidis. BMC Vet Res, 2019. 15(1): p. 319.
- Dhakal, B.K. and M.A. Mulvey, The UPEC pore-forming toxin α-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host Microbe, 2012. 11(1): p. 58-69.
- Ristow, L.C. and R.A. Welch, Hemolysin of uropathogenic Escherichia coli: A cloak or a dagger? Biochim Biophys Acta, 2016. 1858(3): p. 538-45.
- I Gómez, M.; Lee, A.; Reddy, B.; Muir, A.; Soong, G.; Pitt, A.; Cheung, A.; Prince, A. Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat. Med. 2004, 10, 842–848. [Google Scholar] [CrossRef]
- Carbonetti, N.H. Pertussis toxin and adenylate cyclase toxin: key virulence factors of Bordetella pertussis and cell biology tools. Future Microbiol. 2010, 5, 455–469. [Google Scholar] [CrossRef]
- Michalska, M.; Wolf, P. Pseudomonas Exotoxin A: optimized by evolution for effective killing. Front. Microbiol. 2015, 6, 963. [Google Scholar] [CrossRef]
- Di Bella, S., et al., Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins (Basel), 2016. 8(5).
- Melton-Celsa, A.R. Shiga Toxin (Stx) Classification, Structure, and Function. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]
- Ruggenenti, P., M. Noris, and G. Remuzzi, Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney International, 2001. 60(3): p. 831-846.
- Rodríguez-Rubio, L.; Haarmann, N.; Schwidder, M.; Muniesa, M.; Schmidt, H. Bacteriophages of Shiga Toxin-Producing Escherichia coli and Their Contribution to Pathogenicity. Pathogens 2021, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Neely, M.N.; Zhang, X.; Acheson, D.W.K.; Waldor, M.K.; Friedman, D.I. Role for a Phage Promoter in Shiga Toxin 2 Expression from a Pathogenic Escherichia coli Strain. J. Bacteriol. 2001, 183, 2081–2085. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.B.; Mekalanos, J.J. Iron regulation of Shiga-like toxin expression in Escherichia coli is mediated by the fur locus. J. Bacteriol. 1987, 169, 4759–4764. [Google Scholar] [CrossRef]
- Melson, E.M. and M.M. Kendall, The sRNA DicF integrates oxygen sensing to enhance enterohemorrhagic Escherichia coli virulence via distinctive RNA control mechanisms. Proceedings of the National Academy of Sciences, 2019. 116(28): p. 14210-14215.
- Wilke, G.A. and J.B. Wardenburg, Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus α-hemolysin–mediated cellular injury. Proceedings of the National Academy of Sciences, 2010. 107(30): p. 13473-13478.
- Tam, K.; Torres, V.J. Staphylococcus aureusSecreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci—an overview. Front. Microbiol. 2015, 6, 1174. [Google Scholar] [CrossRef]
- Boisset, S.; Geissmann, T.; Huntzinger, E.; Fechter, P.; Bendridi, N.; Possedko, M.; Chevalier, C.; Helfer, A.C.; Benito, Y.; Jacquier, A.; et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev. 2007, 21, 1353–1366. [Google Scholar] [CrossRef]
- Gupta, R.K.; Luong, T.T.; Lee, C.Y. RNAIII of the Staphylococcus aureus agr system activates global regulator MgrA by stabilizing mRNA. Proc. Natl. Acad. Sci. 2015, 112, 14036–14041. [Google Scholar] [CrossRef]
- Le Huyen, K.B.; Gonzalez, C.D.; Pascreau, G.; Bordeau, V.; Cattoir, V.; Liu, W.; Bouloc, P.; Felden, B.; Chabelskaya, S. A small regulatory RNA alters Staphylococcus aureus virulence by titrating RNAIII activity. Nucleic Acids Res. 2021, 49, 10644–10656. [Google Scholar] [CrossRef]
- Morales-Filloy, H.G.; Zhang, Y.; Nübel, G.; George, S.E.; Korn, N.; Wolz, C.; Jäschke, A. The 5′ NAD Cap of RNAIII Modulates Toxin Production in Staphylococcus aureus Isolates. J. Bacteriol. 2020, 202. [Google Scholar] [CrossRef]
- Wiedermannová, J.; Babu, R.; Yuzenkova, Y. Stochastic nature and physiological implications of 5′-NAD RNA cap in bacteria. Nucleic Acids Res. 2024, 52, 11838–11852. [Google Scholar] [CrossRef]
- Peng, D.; Luo, X.; Zhang, N.; Guo, S.; Zheng, J.; Chen, L.; Sun, M. Small RNA-mediated Cry toxin silencing allows Bacillus thuringiensis to evade Caenorhabditis elegans avoidance behavioral defenses. Nucleic Acids Res. 2017, 46, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Porcheron, G.; Habib, R.; Houle, S.; Caza, M.; Lépine, F.; Daigle, F.; Massé, E.; Dozois, C.M. The Small RNA RyhB Contributes to Siderophore Production and Virulence of Uropathogenic Escherichia coli. Infect. Immun. 2014, 82, 5056–5068. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Chen, Y.; Wang, P.; He, M.; Shi, Y.; Lai, Y.; Zhu, G.; Wang, H. RyhB in Avian Pathogenic Escherichia coli Regulates the Expression of Virulence-Related Genes and Contributes to Meningitis Development in a Mouse Model. Int. J. Mol. Sci. 2022, 23, 15532. [Google Scholar] [CrossRef] [PubMed]
- Baussier, C., et al., Small RNA OxyS induces resistance to aminoglycosides during oxidative stress by controlling Fe–S cluster biogenesis in Escherichia coli. Proceedings of the National Academy of Sciences, 2024. 121(46): p. e2317858121.
- Mey, A.R.; Craig, S.A.; Payne, S.M. Characterization of Vibrio cholerae RyhB: the RyhB Regulon and Role of ryhB in Biofilm Formation. Infect. Immun. 2005, 73, 5706–5719. [Google Scholar] [CrossRef]
- Matsuura, M. Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front. Immunol. 2013, 4, 109. [Google Scholar] [CrossRef]
- Sansonetti, P.J. and J.P. Di Santo, Debugging how Bacteria Manipulate the Immune Response. Immunity, 2007. 26(2): p. 149-161.
- Wallden, K.; Rivera-Calzada, A.; Waksman, G. Microreview: Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 2010, 12, 1203–1212. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Boumart, Z.; Velge, P.; Wiedemann, A. Multiple invasion mechanisms and different intracellular Behaviors: a new vision ofSalmonella-host cell interaction. FEMS Microbiol. Lett. 2014, 361, 1–7. [Google Scholar] [CrossRef]
- Robbins, J.R.; Barth, A.I.; Marquis, H.; de Hostos, E.L.; Nelson, W.J.; Theriot, J.A. Listeria monocytogenes Exploits Normal Host Cell Processes to Spread from Cell to Cell✪. J. Cell Biol. 1999, 146, 1333–1350. [Google Scholar] [CrossRef]
- Lalaouna, D.; Baude, J.; Wu, Z.; Tomasini, A.; Chicher, J.; Marzi, S.; Vandenesch, F.; Romby, P.; Caldelari, I.; Moreau, K. RsaC sRNA modulates the oxidative stress response of Staphylococcus aureus during manganese starvation. Nucleic Acids Res. 2019, 47, 9871–9887. [Google Scholar] [CrossRef]
- Cunningham, M.W. Pathogenesis of Group A Streptococcal Infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef]
- Cunningham, M.W. Pathogenesis of group A streptococcal infections and their sequelae. Adv Exp Med Biol 2008, 609, 29–42. [Google Scholar]
- Danger, J.L.; Cao, T.N.; Cao, T.H.; Sarkar, P.; Treviño, J.; Pflughoeft, K.J.; Sumby, P. The small regulatory RNA FasX enhances group A Streptococcus virulence and inhibits pilus expression via serotype-specific targets. Mol. Microbiol. 2015, 96, 249–262. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Treviño, J.; Liu, Z.; Perez, N.; Sumby, P. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol. Microbiol. 2010, 78, 1332–1347. [Google Scholar] [CrossRef]
- Martini, B.A., et al., Small RNA MTS1338 Configures a Stress Resistance Signature in Mycobacterium tuberculosis. Int J Mol Sci, 2023. 24(9).
- Arnvig, K.; Young, D. Non-coding RNA and its potential role in Mycobacterium tuberculosis pathogenesis. RNA Biol. 2012, 9, 427–436. [Google Scholar] [CrossRef]
- Moores, A.; Riesco, A.B.; Schwenk, S.; Arnvig, K.B. Expression, maturation and turnover of DrrS, an unusually stable, DosR regulated small RNA in Mycobacterium tuberculosis. PLOS ONE 2017, 12, e0174079. [Google Scholar] [CrossRef]
- Salina, E.G.; Grigorov, A.; Skvortsova, Y.; Majorov, K.; Bychenko, O.; Ostrik, A.; Logunova, N.; Ignatov, D.; Kaprelyants, A.; Apt, A.; et al. MTS1338, A Small Mycobacterium tuberculosis RNA, Regulates Transcriptional Shifts Consistent With Bacterial Adaptation for Entering Into Dormancy and Survival Within Host Macrophages. Front. Cell. Infect. Microbiol. 2019, 9, 405. [Google Scholar] [CrossRef]
- Banerjee, R.; Rudra, P.; Prajapati, R.K.; Sengupta, S.; Mukhopadhyay, J. Optimization of recombinant Mycobacterium tuberculosis RNA polymerase expression and purification. Tuberculosis 2014, 94, 397–404. [Google Scholar] [CrossRef]
- Banerjee, R.; Rudra, P.; Saha, A.; Mukhopadhyay, J. Recombinant Reporter Assay Using Transcriptional Machinery of Mycobacterium tuberculosis. J. Bacteriol. 2014, 197, 646–653. [Google Scholar] [CrossRef]
- Sharma, S., et al., Functional insights into Mycobacterium tuberculosis DevR-dependent transcriptional machinery utilizing Escherichia coli. Biochemical Journal, 2021. 478(16): p. 3079-3098.
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Horrocks, V.; King, O.G.; Yip, A.Y.G.; Marques, I.M.; McDonald, J.A.K. Role of the gut microbiota in nutrient competition and protection against intestinal pathogen colonization. Microbiology 2023, 169, 001377. [Google Scholar] [CrossRef]
- Nguyen, J., J. Lara-Gutiérrez, and R. Stocker, Environmental fluctuations and their effects on microbial communities, populations and individuals. FEMS Microbiol Rev, 2021. 45(4).
- Cox, G.M., et al., Superoxide dismutase influences the virulence of Cryptococcus neoformans by affecting growth within macrophages. Infect Immun, 2003. 71(1): p. 173-80.
- Carlioz, A. and D. Touati, Isolation of superoxide dismutase mutants in Escherichia coli: is superoxide dismutase necessary for aerobic life? Embo j, 1986. 5(3): p. 623-30.
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 1–19. [Google Scholar] [CrossRef]
- Rocha, E.R.; Smith, C.J. Role of the Alkyl Hydroperoxide Reductase ( ahpCF ) Gene in Oxidative Stress Defense of the Obligate Anaerobe Bacteroides fragilis. J. Bacteriol. 1999, 181, 5701–5710. [Google Scholar] [CrossRef]
- Lund, P.A.; De Biase, D.; Liran, O.; Scheler, O.; Mira, N.P.; Cetecioglu, Z.; Fernández, E.N.; Bover-Cid, S.; Hall, R.; Sauer, M.; et al. Understanding How Microorganisms Respond to Acid pH Is Central to Their Control and Successful Exploitation. Front. Microbiol. 2020, 11, 556140. [Google Scholar] [CrossRef]
- Konieczna, I.; Zarnowiec, P.; Kwinkowski, M.; Kolesinska, B.; Fraczyk, J.; Kaminski, Z.; Kaca, W. Bacterial Urease and its Role in Long-Lasting Human Diseases. Curr. Protein Pept. Sci. 2012, 13, 789–806. [Google Scholar] [CrossRef]
- Alteri, C.J.; Mobley, H.L. Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr. Opin. Microbiol. 2012, 15, 3–9. [Google Scholar] [CrossRef]
- Massé, E.; Vanderpool, C.K.; Gottesman, S. Effect of RyhB Small RNA on Global Iron Use in Escherichia coli. J. Bacteriol. 2005, 187, 6962–6971. [Google Scholar] [CrossRef]
- Calderón, P.F.; Morales, E.H.; Acuña, L.G.; Fuentes, D.N.; Gil, F.; Porwollik, S.; McClelland, M.; Saavedra, C.P.; Calderón, I.L. The small RNA RyhB homologs from Salmonella typhimurium participate in the response to S-nitrosoglutathione-induced stress. Biochem. Biophys. Res. Commun. 2014, 450, 641–645. [Google Scholar] [CrossRef]
- Banerjee, R.; Weisenhorn, E.; Schwartz, K.J.; Myers, K.S.; Glasner, J.D.; Perna, N.T.; Coon, J.J.; Welch, R.A.; Kiley, P.J. Tailoring a Global Iron Regulon to a Uropathogen. mBio 2020, 11. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, S.; Wu, N.; Yuan, Y.; Zhang, W.; Zhang, Y. Small Non-coding RNA RyhB Mediates Persistence to Multiple Antibiotics and Stresses in Uropathogenic Escherichia coli by Reducing Cellular Metabolism. Front. Microbiol. 2018, 9, 136. [Google Scholar] [CrossRef]
- Acuña, L.G.; Barros, M.J.; Montt, F.; Peñaloza, D.; Núñez, P.; Valdés, I.; Gil, F.; Fuentes, J.A.; Calderón, I.L. Participation of two sRNA RyhB homologs from the fish pathogen Yersinia ruckeri in bacterial physiology. Microbiol. Res. 2020, 242, 126629. [Google Scholar] [CrossRef]
- Coronel-Tellez, R.H., et al., sRNA-controlled iron sparing response in Staphylococci. Nucleic Acids Res, 2022. 50(15): p. 8529-8546.
- Gerrick, E.R.; Barbier, T.; Chase, M.R.; Xu, R.; François, J.; Lin, V.H.; Szucs, M.J.; Rock, J.M.; Ahmad, R.; Tjaden, B.; et al. Small RNA profiling in Mycobacterium tuberculosis identifies MrsI as necessary for an anticipatory iron sparing response. Proc. Natl. Acad. Sci. 2018, 115, 6464–6469. [Google Scholar] [CrossRef] [PubMed]
- Girardin, R.C.; McDonough, K.A. Small RNA Mcr11 requires the transcription factor AbmR for stable expression and regulates genes involved in the central metabolism of Mycobacterium tuberculosis. Mol. Microbiol. 2019, 113, 504–520. [Google Scholar] [CrossRef]
- Rudra, P., et al., Novel mechanism of gene regulation: the protein Rv1222 of Mycobacterium tuberculosis inhibits transcription by anchoring the RNA polymerase onto DNA. Nucleic Acids Research, 2015. 43(12): p. 5855-5867.
- Altuvia, S.; Weinstein-Fischer, D.; Zhang, A.; Postow, L.; Storz, G. A Small, Stable RNA Induced by Oxidative Stress: Role as a Pleiotropic Regulator and Antimutator. Cell 1997, 90, 43–53. [Google Scholar] [CrossRef]
- Åslund, F., et al., Regulation of the OxyR transcription factor by hydrogen peroxide and the cellular thiol—disulfide status. Proceedings of the National Academy of Sciences, 1999. 96(11): p. 6161-6165.
- Štih, V., et al., Spatial arrangement of functional domains in OxyS stress response sRNA. Rna, 2023. 29(10): p. 1520-1534.
- Soper, T.; Mandin, P.; Majdalani, N.; Gottesman, S.; Woodson, S.A. Positive regulation by small RNAs and the role of Hfq. Proc. Natl. Acad. Sci. 2010, 107, 9602–9607. [Google Scholar] [CrossRef]
- Barshishat, S.; Elgrably-Weiss, M.; Edelstein, J.; Georg, J.; Govindarajan, S.; Haviv, M.; Wright, P.R.; Hess, W.R.; Altuvia, S. OxyS small RNA induces cell cycle arrest to allow DNA damage repair. EMBO J. 2017, 37, 413–426. [Google Scholar] [CrossRef]
- Cho, H.; Kim, K.-S. Escherichia coli OxyS RNA triggers cephalothin resistance by modulating the expression of CRP-associated genes. Biochem. Biophys. Res. Commun. 2018, 506, 66–72. [Google Scholar] [CrossRef]
- Sledjeski, D.D.; Gupta, A.; Gottesman, S. The small RNA, DsrA, is essential for the low temperature expression of RpoS during exponential growth in Escherichia coli. EMBO J. 1996, 15, 3993–4000. [Google Scholar] [CrossRef]
- Lease, R.A.; Smith, D.; McDonough, K.; Belfort, M. The Small Noncoding DsrA RNA Is an Acid Resistance Regulator inEscherichia coli. J. Bacteriol. 2004, 186, 6179–6185. [Google Scholar] [CrossRef]
- Dong, R.; Liang, Y.; He, S.; Cui, Y.; Shi, C.; He, Y.; Shi, X. DsrA Modulates Central Carbon Metabolism and Redox Balance by Directly Repressing pflB Expression in Salmonella Typhimurium. Microbiol. Spectr. 2022, 10, e0152221. [Google Scholar] [CrossRef]
- Sledjeski, D.; Gottesman, S. A small RNA acts as an antisilencer of the H-NS-silenced rcsA gene of Escherichia coli. Proc. Natl. Acad. Sci. 1995, 92, 2003–2007. [Google Scholar] [CrossRef]
- Rozanov, D.V., R. D'Ari, and S.P. Sineoky, RecA-independent pathways of lambdoid prophage induction in Escherichia coli. J Bacteriol, 1998. 180(23): p. 6306-15.
- Zhu, W.; Xi, L.; Qiao, J.; Du, D.; Wang, Y. ; Morigen Involvement of OxyR and Dps in the repression of replication initiation by DsrA small RNA in Escherichia coli. Gene 2023, 882, 147659. [Google Scholar] [CrossRef] [PubMed]
- Ades, S.E., Regulation by destruction: design of the sigmaE envelope stress response. Curr Opin Microbiol, 2008. 11(6): p. 535-40.
- Thompson, K.M., V.A. Rhodius, and S. Gottesman, SigmaE regulates and is regulated by a small RNA in Escherichia coli. J Bacteriol, 2007. 189(11): p. 4243-56.
- Papenfort, K.; Bouvier, M.; Mika, F.; Sharma, C.M.; Vogel, J. Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc. Natl. Acad. Sci. 2010, 107, 20435–20440. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.P.; Baniulyte, G.; Esnault, C.; Chegireddy, K.; Singh, N.; Monge, M.; Dale, R.K.; Storz, G.; Wade, J.T. Regulatory roles of Escherichia coli 5' UTR and ORF-internal RNAs detected by 3' end mapping. eLife 2021, 10. [Google Scholar] [CrossRef]
- Aminov, R.I. A Brief History of the Antibiotic Era: Lessons Learned and Challenges for the Future. Front. Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef]
- Hutchings, M.I., A.W. Truman, and B. Wilkinson, Antibiotics: past, present and future. Current Opinion in Microbiology, 2019. 51: p. 72-80.
- Krause, K.M., et al., Aminoglycosides: An Overview. Cold Spring Harb Perspect Med, 2016. 6(6).
- Kounatidis, D., et al., Third-Generation Tetracyclines: Current Knowledge and Therapeutic Potential. Biomolecules, 2024. 14(7).
- Bosnar, M.; Kelnerić, Z.; Munić, V.; Eraković, V.; Parnham, M.J. Cellular Uptake and Efflux of Azithromycin, Erythromycin, Clarithromycin, Telithromycin, and Cethromycin. Antimicrob. Agents Chemother. 2005, 49, 2372–2377. [Google Scholar] [CrossRef]
- Hooper, D.C.; Jacoby, G.A. Topoisomerase Inhibitors: Fluoroquinolone Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025320. [Google Scholar] [CrossRef]
- Ovung, A.; Bhattacharyya, J. Sulfonamide drugs: structure, antibacterial property, toxicity, and biophysical interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef]
- Zeng, D.; Debabov, D.; Hartsell, T.L.; Cano, R.J.; Adams, S.; Schuyler, J.A.; McMillan, R.; Pace, J.L. Approved Glycopeptide Antibacterial Drugs: Mechanism of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a026989. [Google Scholar] [CrossRef] [PubMed]
- Howden, B.P.; Beaume, M.; Harrison, P.F.; Hernandez, D.; Schrenzel, J.; Seemann, T.; Francois, P.; Stinear, T.P. Analysis of the Small RNA Transcriptional Response in Multidrug-Resistant Staphylococcus aureus after Antimicrobial Exposure. Antimicrob. Agents Chemother. 2013, 57, 3864–3874. [Google Scholar] [CrossRef]
- Molina-Santiago, C., et al., Differential transcriptional response to antibiotics by Pseudomonas putida DOT-T1E. Environ Microbiol, 2015. 17(9): p. 3251-62.
- Jin, Y.; Watt, R.M.; Danchin, A.; Huang, J.-D. Small noncoding RNA GcvB is a novel regulator of acid resistance in Escherichia coli. BMC Genom. 2009, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Goto, S.; Kurita, D.; Ushida, C.; Himeno, H. Involvement of GcvB small RNA in intrinsic resistance to multiple aminoglycoside antibiotics in Escherichia coli. J. Biochem. 2020, 169, 485–489. [Google Scholar] [CrossRef]
- Liu, B.; Fang, J.; Chen, H.; Sun, Y.; Yang, S.; Gao, Q.; Zhang, Y.; Chen, C. GcvB Regulon Revealed by Transcriptomic and Proteomic Analysis in Vibrio alginolyticus. Int. J. Mol. Sci. 2022, 23, 9399. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, M.; Okayama, H.; Lejars, M.; Kanda, T.; Tanaka, Y.; Itaya, K.; Okuno, M.; Itoh, T.; Iwai, N.; Wachi, M. Mining RNA-seq data reveals the massive regulon of GcvB small RNA and its physiological significance in maintaining amino acid homeostasis in Escherichia coli. Mol. Microbiol. 2021, 117, 160–178. [Google Scholar] [CrossRef] [PubMed]
- Miyakoshi, M., Y. Chao, and J. Vogel, Cross talk between ABC transporter mRNAs via a target mRNA-derived sponge of the GcvB small RNA. Embo j, 2015. 34(11): p. 1478-92.
- Lalaouna, D.; Eyraud, A.; Devinck, A.; Prévost, K.; Massé, E. GcvB small RNA uses two distinct seed regions to regulate an extensive targetome. Mol. Microbiol. 2018, 111, 473–486. [Google Scholar] [CrossRef]
- Stauffer, L.T.; Stauffer, G.V. The Escherichia coli GcvB sRNA Uses Genetic Redundancy to Control cycA Expression. ISRN Microbiol. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Barreto, B.; Rogers, E.; Xia, J.; Frisch, R.L.; Richters, M.; Fitzgerald, D.M.; Rosenberg, S.M. The Small RNA GcvB Promotes Mutagenic Break Repair by Opposing the Membrane Stress Response. J. Bacteriol. 2016, 198, 3296–3308. [Google Scholar] [CrossRef]
- Delihas, N.; Forst, S. MicF : an antisense RNA gene involved in response of Escherichia coli to global stress factors 1 1Edited by D. Draper. J. Mol. Biol. 2001, 313, 1–12. [Google Scholar] [CrossRef]
- Cowan, S.W.; Garavito, R.M.; Jansonius, J.N.; Jenkins, J.A.; Karlsson, R.; König, N.; Pai, E.F.; Pauptit, R.A.; Rizkallah, P.; Rosenbusch, J.; et al. The structure of OmpF porin in a tetragonal crystal form. Structure 1995, 3, 1041–1050. [Google Scholar] [CrossRef]
- Carrier, M.-C.; Lalaouna, D.; Massé, E. Hfq protein and GcvB small RNA tailoring of oppA target mRNA to levels allowing translation activation by MicF small RNA in Escherichia coli. RNA Biol. 2023, 20, 59–76. [Google Scholar] [CrossRef]
- Olaitan, A.O.; Morand, S.; Rolain, J.-M. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-ResistantPseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef] [PubMed]
- Ocampo-Sosa, A.A.; Cabot, G.; Rodríguez, C.; Roman, E.; Tubau, F.; Macia, M.D.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; et al. Alterations of OprD in Carbapenem-Intermediate and -Susceptible Strains of Pseudomonas aeruginosa Isolated from Patients with Bacteremia in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 2012, 56, 1703–1713. [Google Scholar] [CrossRef]
- Zhang, Y.F., et al., Probing the sRNA regulatory landscape of P. aeruginosa: post-transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol Microbiol, 2017. 106(6): p. 919-937.
- Sonnleitner, E.; Pusic, P.; Wolfinger, M.T.; Bläsi, U. Distinctive Regulation of Carbapenem Susceptibility in Pseudomonas aeruginosa by Hfq. Front. Microbiol. 2020, 11, 1001. [Google Scholar] [CrossRef]
- Schumann, A.; Gaballa, A.; Wiedmann, M. The multifaceted roles of phosphoethanolamine-modified lipopolysaccharides: from stress response and virulence to cationic antimicrobial resistance. Microbiol. Mol. Biol. Rev. 2024, 88, e0019323. [Google Scholar] [CrossRef]
- Wang, J.; Ma, W.; Fang, Y.; Liang, H.; Yang, H.; Wang, Y.; Dong, X.; Zhan, Y.; Wang, X. Core Oligosaccharide Portion of Lipopolysaccharide Plays Important Roles in Multiple Antibiotic Resistance in Escherichia coli. Antimicrob. Agents Chemother. 2021, 65, AAC0034121. [Google Scholar] [CrossRef]
- Moon, K.; Six, D.A.; Lee, H.; Raetz, C.R.H.; Gottesman, S. Complex transcriptional and post-transcriptional regulation of an enzyme for lipopolysaccharide modification. Mol. Microbiol. 2013, 89, 52–64. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Cui, L.; Kuroda, M.; Ito, T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001, 9, 486–493. [Google Scholar] [CrossRef]
- Bohn, C.; Rigoulay, C.; Chabelskaya, S.; Sharma, C.M.; Marchais, A.; Skorski, P.; Borezée-Durant, E.; Barbet, R.; Jacquet, E.; Jacq, A.; et al. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res. 2010, 38, 6620–6636. [Google Scholar] [CrossRef]
- Kathirvel, M.; Buchad, H.; Nair, M. Enhancement of the pathogenicity of Staphylococcus aureus strain Newman by a small noncoding RNA SprX1. Med Microbiol. Immunol. 2016, 205, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Eyraud, A.; Tattevin, P.; Chabelskaya, S.; Felden, B. A small RNA controls a protein regulator involved in antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res. 2014, 42, 4892–4905. [Google Scholar] [CrossRef]
- Ivain, L.; Bordeau, V.; Eyraud, A.; Hallier, M.; Dreano, S.; Tattevin, P.; Felden, B.; Chabelskaya, S. An in vivo reporter assay for sRNA-directed gene control in Gram-positive bacteria: identifying a novel sRNA target in Staphylococcus aureus. Nucleic Acids Res. 2017, 45, 4994–5007. [Google Scholar] [CrossRef]
- Buchad, H.; Nair, M. The small RNA SprX regulates the autolysin regulator WalR in Staphylococcus aureus. Microbiol. Res. 2021, 250, 126785. [Google Scholar] [CrossRef]
- Wurtzel, O.; Yoder-Himes, D.R.; Han, K.; Dandekar, A.A.; Edelheit, S.; Greenberg, E.P.; Sorek, R.; Lory, S. The Single-Nucleotide Resolution Transcriptome of Pseudomonas aeruginosa Grown in Body Temperature. PLOS Pathog. 2012, 8, e1002945. [Google Scholar] [CrossRef]
- Di Maso, A.M. and C. Ruiz, Physiological Effects of TolC-Dependent Multidrug Efflux Pumps in Escherichia coli: Impact on Motility and Growth Under Stress Conditions. MicrobiologyOpen, 2024. 13(6): p. e70006.
- Du, D., et al., Structure of the AcrAB-TolC multidrug efflux pump. Nature, 2014. 509(7501): p. 512-5.
- Smith, B.L., S. Fernando, and M.D. King, Escherichia coli resistance mechanism AcrAB-TolC efflux pump interactions with commonly used antibiotics: a molecular dynamics study. Scientific Reports, 2024. 14(1): p. 2742.
- Fitzpatrick, A.W.P.; Llabrés, S.; Neuberger, A.; Blaza, J.N.; Bai, X.-C.; Okada, U.; Murakami, S.; van Veen, H.W.; Zachariae, U.; Scheres, S.H.W.; et al. Structure of the MacAB–TolC ABC-type tripartite multidrug efflux pump. Nat. Microbiol. 2017, 2, 1–8. [Google Scholar] [CrossRef]
- Horiyama, T.; Nishino, K. AcrB, AcrD, and MdtABC Multidrug Efflux Systems Are Involved in Enterobactin Export in Escherichia coli. PLOS ONE 2014, 9, e108642. [Google Scholar] [CrossRef]
- Yousefian, N., et al., Structural characterization of the EmrAB-TolC efflux complex from E. coli. Biochim Biophys Acta Biomembr, 2021. 1863(1): p. 183488.
- Muteeb, G.; Rehman, T.; Shahwan, M.; Aatif, M. Origin of Antibiotics and Antibiotic Resistance, and Their Impacts on Drug Development: A Narrative Review. Pharmaceuticals 2023, 16, 1615. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef]
- Zhang, H.; Song, T.; Qin, C.; Xu, H.; Qiao, M. A Novel Non-Coding RNA CsiR Regulates the Ciprofloxacin Resistance in Proteus vulgaris by Interacting with emrB mRNA. Int. J. Mol. Sci. 2021, 22, 10627. [Google Scholar] [CrossRef]
- Coleman, S.R.; Smith, M.L.; Spicer, V.; Lao, Y.; Mookherjee, N.; Hancock, R.E.W. Overexpression of the Small RNA PA0805.1 in Pseudomonas aeruginosa Modulates the Expression of a Large Set of Genes and Proteins, Resulting in Altered Motility, Cytotoxicity, and Tobramycin Resistance. mSystems 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S., et al., The small RNA, SdsR, acts as a novel type of toxin in Escherichia coli. RNA Biol, 2018. 15(10): p. 1319-1335.
- Parker, A.; Gottesman, S. Small RNA Regulation of TolC, the Outer Membrane Component of Bacterial Multidrug Transporters. J. Bacteriol. 2016, 198, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Laureti, L.; Crussard, S.; Abida, H.; Rodríguez-Rojas, A.; Blazquez, J.; Baharoglu, Z.; Mazel, D.; Darfeuille, F.; Vogel, J.; et al. β-lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat. Commun. 2013, 4, 1610. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar] [CrossRef]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef]
- Karygianni, L.; Ren, Z.; Koo, H.; Thurnheer, T. Biofilm Matrixome: Extracellular Components in Structured Microbial Communities. Trends Microbiol. 2020, 28, 668–681. [Google Scholar] [CrossRef]
- Waters, E.M., et al., Convergence of Staphylococcus aureus Persister and Biofilm Research: Can Biofilms Be Defined as Communities of Adherent Persister Cells? PLoS Pathog, 2016. 12(12): p. e1006012.
- Sultan, M.; Arya, R.; Kim, K.K. Roles of Two-Component Systems in Pseudomonas aeruginosa Virulence. Int. J. Mol. Sci. 2021, 22, 12152. [Google Scholar] [CrossRef]
- Mukherjee, S.; Moustafa, D.; Smith, C.D.; Goldberg, J.B.; Bassler, B.L. The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer. PLOS Pathog. 2017, 13, e1006504. [Google Scholar] [CrossRef]
- Van Puyvelde, S.; Steenackers, H.P.; Vanderleyden, J. Small RNAs regulating biofilm formation and outer membrane homeostasis. RNA Biol. 2013, 10, 185–191. [Google Scholar] [CrossRef]
- Timmermans, J. and L. Van Melderen, Post-transcriptional global regulation by CsrA in bacteria. Cell Mol Life Sci, 2010. 67(17): p. 2897-908.
- Liu, P.; Yue, C.; Liu, L.; Gao, C.; Lyu, Y.; Deng, S.; Tian, H.; Jia, X. The function of small RNA in Pseudomonas aeruginosa. PeerJ 2022, 10, e13738. [Google Scholar] [CrossRef] [PubMed]
- Law, C.O.K.; Huang, C.; Pan, Q.; Lee, J.; Hao, Q.; Chan, T.-F.; Lo, N.W.S.; Ang, I.L.; Koon, A.; Ip, M.; et al. A Small RNA Transforms the Multidrug Resistance of Pseudomonas aeruginosa to Drug Susceptibility. Mol. Ther. - Nucleic Acids 2019, 16, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y., et al., A phosphate starvation induced small RNA promotes Bacillus biofilm formation. npj Biofilms and Microbiomes, 2024. 10(1): p. 115.
- Taylor, P.K.; Van Kessel, A.T.M.; Colavita, A.; Hancock, R.E.W.; Mah, T.-F. A novel small RNA is important for biofilm formation and pathogenicity in Pseudomonas aeruginosa. PLOS ONE 2017, 12, e0182582–e0182582. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Waters, C.M.; Bassler, B.L. QUORUM SENSING: Cell-to-Cell Communication in Bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef]
- Wu, L.; Luo, Y. Bacterial Quorum-Sensing Systems and Their Role in Intestinal Bacteria-Host Crosstalk. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Smith, R.S. and B.H. Iglewski, Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest, 2003. 112(10): p. 1460-5.
- Yarwood, J.M. and P.M. Schlievert, Quorum sensing in Staphylococcus infections. J Clin Invest, 2003. 112(11): p. 1620-5.
- Zhu, J.; Mekalanos, J.J. Quorum Sensing-Dependent Biofilms Enhance Colonization in Vibrio cholerae. Dev. Cell 2003, 5, 647–656. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Ding, T. Quorum-Sensing Regulation of Antimicrobial Resistance in Bacteria. Microorganisms 2020, 8, 425. [Google Scholar] [CrossRef]
- Anetzberger, C.; Reiger, M.; Fekete, A.; Schell, U.; Stambrau, N.; Plener, L.; Kopka, J.; Schmitt-Kopplin, P.; Hilbi, H.; Jung, K. Autoinducers Act as Biological Timers in Vibrio harveyi. PLOS ONE 2012, 7, e48310. [Google Scholar] [CrossRef]
- Waters, C.M.; Bassler, B.L. The Vibrio harveyi quorum-sensing system uses shared regulatory components to discriminate between multiple autoinducers. Genes Dev. 2006, 20, 2754–2767. [Google Scholar] [CrossRef]
- Mok, K.C.; Wingreen, N.S.; Bassler, B.L. Vibrio harveyi quorum sensing: a coincidence detector for two autoinducers controls gene expression. EMBO J. 2003, 22, 870–881. [Google Scholar] [CrossRef]
- Papenfort, K.; E Silpe, J.; Schramma, K.R.; Cong, J.-P.; Seyedsayamdost, M.R.; Bassler, B.L. A Vibrio cholerae autoinducer–receptor pair that controls biofilm formation. Nat. Chem. Biol. 2017, 13, 551–557. [Google Scholar] [CrossRef]
- Jemielita, M.; Wingreen, N.S.; Bassler, B.L.; States, U. Quorum sensing controls Vibrio cholerae multicellular aggregate formation. eLife 2018, 7. [Google Scholar] [CrossRef]
- Tsou, A.M.; Liu, Z.; Cai, T.; Zhu, J. The VarS/VarA two-component system modulates the activity of the Vibrio cholerae quorum-sensing transcriptional regulator HapR. Microbiology 2011, 157, 1620–1628. [Google Scholar] [CrossRef]
- Shao, Y. and B.L. Bassler, Quorum regulatory small RNAs repress type VI secretion in ibrio cholerae. Molecular Microbiology, 2014. 92(5): p. 921-930.
- MacIntyre, D.L.; Miyata, S.T.; Kitaoka, M.; Pukatzki, S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc. Natl. Acad. Sci. 2010, 107, 19520–19524. [Google Scholar] [CrossRef]
- Allsopp, L.P.; Bernal, P. Killing in the name of: T6SS structure and effector diversity. Microbiology 2023, 169, 001367. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.G.; Ho, B.T.; Yoder-Himes, D.R.; Mekalanos, J.J. Identification of T6SS-dependent effector and immunity proteins by Tn-seq in Vibrio cholerae. Proc. Natl. Acad. Sci. 2013, 110, 2623–2628. [Google Scholar] [CrossRef]
- Hersch, S.J.; Manera, K.; Dong, T.G. Defending against the Type Six Secretion System: beyond Immunity Genes. Cell Rep. 2020, 33, 108259. [Google Scholar] [CrossRef]
- Huber, M.; Lippegaus, A.; Melamed, S.; Siemers, M.; Wucher, B.R.; Hoyos, M.; Nadell, C.; Storz, G.; Papenfort, K. An RNA sponge controls quorum sensing dynamics and biofilm formation in Vibrio cholerae. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef]
- Smith, R.S.; Iglewski, B.H. P. aeruginosa quorum-sensing systems and virulence. Curr. Opin. Microbiol. 2003, 6, 56–60. [Google Scholar] [CrossRef] [PubMed]
- McKnight, S.L., B.H. Iglewski, and E.C. Pesci, The Pseudomonas quinolone signal regulates rhl quorum sensing in Pseudomonas aeruginosa. J Bacteriol, 2000. 182(10): p. 2702-8.
- Jimenez, P.N.; Koch, G.; Thompson, J.A.; Xavier, K.B.; Cool, R.H.; Quax, W.J. The Multiple Signaling Systems Regulating Virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 2012, 76, 46–65. [Google Scholar] [CrossRef]
- Managò, A., et al., Pseudomonas aeruginosa pyocyanin induces neutrophil death via mitochondrial reactive oxygen species and mitochondrial acid sphingomyelinase. Antioxid Redox Signal, 2015. 22(13): p. 1097-110.
- Sonnleitner, E.; Gonzalez, N.; Sorger-Domenigg, T.; Heeb, S.; Richter, A.S.; Backofen, R.; Williams, P.; Hüttenhofer, A.; Haas, D.; Bläsi, U. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 2011, 80, 868–885. [Google Scholar] [CrossRef]
- Carloni, S.; Macchi, R.; Sattin, S.; Ferrara, S.; Bertoni, G. The small RNA ReaL: a novel regulatory element embedded in the Pseudomonas aeruginosa quorum sensing networks. Environ. Microbiol. 2017, 19, 4220–4237. [Google Scholar] [CrossRef]
- Pu, J.; Zhang, S.; He, X.; Zeng, J.; Shen, C.; Luo, Y.; Li, H.; Long, Y.; Liu, J.; Xiao, Q.; et al. The Small RNA AmiL Regulates Quorum Sensing-Mediated Virulence in Pseudomonas aeruginosa PAO1. Microbiol. Spectr. 2022, 10, e0221121. [Google Scholar] [CrossRef]
- Clamens, T.; Rosay, T.; Crépin, A.; Grandjean, T.; Kentache, T.; Hardouin, J.; Bortolotti, P.; Neidig, A.; Mooij, M.; Hillion, M.; et al. The aliphatic amidase AmiE is involved in regulation of Pseudomonas aeruginosa virulence. Sci. Rep. 2017, 7, srep41178. [Google Scholar] [CrossRef]
- Lu, Y., et al., Identification of a novel RhlI/R-PrrH-LasI/Phzc/PhzD signalling cascade and its implication in P. aeruginosa virulence. Emerg Microbes Infect, 2019. 8(1): p. 1658-1667.
- Jia, T.; Bi, X.; Li, M.; Zhang, C.; Ren, A.; Li, S.; Zhou, T.; Zhang, Y.; Liu, Y.; Liu, X.; et al. Hfq-binding small RNA PqsS regulates Pseudomonas aeruginosa pqs quorum sensing system and virulence. npj Biofilms Microbiomes 2024, 10, 1–16. [Google Scholar] [CrossRef]
- Chabelskaya, S.; Gaillot, O.; Felden, B. A Staphylococcus aureus Small RNA Is Required for Bacterial Virulence and Regulates the Expression of an Immune-Evasion Molecule. PLOS Pathog. 2010, 6, e1000927. [Google Scholar] [CrossRef]
- Melamed, S.; Faigenbaum-Romm, R.; Peer, A.; Reiss, N.; Shechter, O.; Bar, A.; Altuvia, Y.; Argaman, L.; Margalit, H. Mapping the small RNA interactome in bacteria using RIL-seq. Nat. Protoc. 2017, 13, 1–33. [Google Scholar] [CrossRef]
- Han, K.; Tjaden, B.; Lory, S. GRIL-seq provides a method for identifying direct targets of bacterial small regulatory RNA by in vivo proximity ligation. Nat. Microbiol. 2016, 2, 16239–16239. [Google Scholar] [CrossRef]
- Boudry, P.; Piattelli, E.; Drouineau, E.; Peltier, J.; Boutserin, A.; Lejars, M.; Hajnsdorf, E.; Monot, M.; Dupuy, B.; Martin-Verstraete, I.; et al. Identification of RNAs bound by Hfq reveals widespread RNA partners and a sporulation regulator in the human pathogen Clostridioides difficile. RNA Biol. 2021, 18, 1931–1952. [Google Scholar] [CrossRef]
- Banerjee, R.; Askenasy, I.; Mettert, E.L.; Kiley, P.J. Iron–sulfur Rrf2 transcription factors: an emerging versatile platform for sensing stress. Curr. Opin. Microbiol. 2024, 82, 102543. [Google Scholar] [CrossRef]
- Balderas, D.; Mettert, E.; Lam, H.N.; Banerjee, R.; Gverzdys, T.; Alvarez, P.; Saarunya, G.; Tanner, N.; Zoubedi, A.; Wei, Y.; et al. Genome Scale Analysis Reveals IscR Directly and Indirectly Regulates Virulence Factor Genes in Pathogenic Yersinia. mBio 2021, 12, e0063321. [Google Scholar] [CrossRef]
- Azaldegui, C.A.; Vecchiarelli, A.G.; Biteen, J.S. The emergence of phase separation as an organizing principle in bacteria. Biophys. J. 2021, 120, 1123–1138. [Google Scholar] [CrossRef]
- Banerjee, R. Synthetic Biology-Based Approaches to Investigate Host–Pathogen Interactions. SynBio 2025, 3, 4. [Google Scholar] [CrossRef]
- Wang, J.; Lu, X.; Wang, C.; Yue, Y.; Wei, B.; Zhang, H.; Wang, H.; Chen, J. Research Progress on the Combination of Quorum-Sensing Inhibitors and Antibiotics against Bacterial Resistance. Molecules 2024, 29, 1674. [Google Scholar] [CrossRef]
- Wang, S.; Zhuang, X.; Dong, W.; Xin, F.; Jia, H.; Wu, X. Advances in the Application of Quorum Sensing to Regulate Electrode Biofilms in Bioelectrochemical Systems. Fermentation 2023, 9, 625. [Google Scholar] [CrossRef]
- Davies, J.; Spiegelman, G.B.; Yim, G. The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 2006, 9, 445–453. [Google Scholar] [CrossRef]
- Yu, J.; Schneiders, T. Tigecycline challenge triggers sRNA production in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2012, 12, 195–195. [Google Scholar] [CrossRef]
- Chen, Y.; et al. Small RNAs in the genus Clostridium. mBio 2011, 2, e00340–10. [Google Scholar] [CrossRef]
- Poonawala, H.; Zhang, Y.; Kuchibhotla, S.; Green, A.G.; Cirillo, D.M.; Di Marco, F.; Spitlaeri, A.; Miotto, P.; Farhat, M.R. Transcriptomic responses to antibiotic exposure in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2024, 68, e0118523. [Google Scholar] [CrossRef] [PubMed]
- Adnan, S.-N.; Ibrahim, N.; Yaacob, W.A. Transcriptome analysis of methicillin-resistant Staphylococcus aureus in response to stigmasterol and lupeol. J. Glob. Antimicrob. Resist. 2017, 8, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Conde, S.; de la Torre, J.; Molina-Santiago, C.; Ramos, J.-L.; Duque, E. Mechanisms of Resistance to Chloramphenicol in Pseudomonas putida KT2440. Antimicrob. Agents Chemother. 2012, 56, 1001–1009. [Google Scholar] [CrossRef]
- Battesti, A.; Majdalani, N.; Gottesman, S. The RpoS-Mediated General Stress Response inEscherichia coli. Annu. Rev. Microbiol. 2011, 65, 189–213. [Google Scholar] [CrossRef]
- Layton, J.C.; Foster, P.L. Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli. Mol Microbiol 2003, 50, 549–61. [Google Scholar] [CrossRef] [PubMed]
- Cen, T.; Zhang, X.; Xie, S.; Li, D. Preservatives accelerate the horizontal transfer of plasmid-mediated antimicrobial resistance genes via differential mechanisms. 2020, 138, 105544. https://doi.org/10.1016/j.envint.2020.105544. [CrossRef]
- Chan, H.; Ho, J.; Liu, X.; Zhang, L.; Wong, S.H.; Chan, M.T.; Wu, W.K. Potential and use of bacterial small RNAs to combat drug resistance: a systematic review. Infect. Drug Resist. 2017, ume 10, 521–532. [Google Scholar] [CrossRef]
- Qian, W.; Sun, J.; Liu, T.; Yang, Z.; Tsui, S.K.-W. sRNAdeep: a novel tool for bacterial sRNA prediction based on DistilBERT encoding mode and deep learning algorithms. BMC Genom. 2024, 25, 1–14. [Google Scholar] [CrossRef]
- Tjaden, B. TargetRNA3: predicting prokaryotic RNA regulatory targets with machine learning. Genome Biol. 2023, 24, 1–13. [Google Scholar] [CrossRef]
Figure 1.
Role of Bacterial sRNAs in Virulence (a) Regulation of P-pili expression by PapR: The expression of P-pili in uropathogenic Escherichia coli (UPEC) is controlled by a phase switch that toggles between the OFF and ON states. Upper left panel: In the OFF state, the phase switch is regulated by the interaction of Lrp, papI, papB, and Dam methylase. The intergenic region between papB and papI contains two sets of methylation sites: proximal and distal. Binding of Lrp to the proximal site prevents Dam methylation, leading to methylation of the distal site and suppression of pap operon transcription, thereby preventing P-fimbriae expression. Upper right panel: In the ON state, Lrp, in association with papI, binds to the proximal site, facilitating activation of the phase switch and allowing transcription of the pap operon, leading to P-fimbriae expression. Lower left panel: The small RNA PapR post-transcriptionally represses papI expression by directly interacting with its transcript, preventing ribosome binding. In the absence of papI, Lrp cannot activate the phase switch, thereby repressing P-fimbriae expression. (b) Structural components of P-fimbriae. A schematic representation of the major structural components of P-fimbriae, which facilitate bacterial adherence to host epithelial cells.(c) Role of StxS in Shiga toxin phage regulation:Left panel (Lysogeny): During lysogeny, the late promoters (PR′) of Stx1 and Stx2 phages are constitutively active, transcribing StxSL, which terminates at the intrinsic terminator tR′. The StxSL transcript is processed by RNase E to generate the regulatory small RNA StxS. StxS enhances the expression of rpoS, the stationary-phase sigma factor, while repressing stx1B, encoding the Shiga toxin 1B subunit, thereby reducing Shiga toxin production. Right panel (Lytic induction): During the lytic cycle, the phage-encoded antiterminator Q is expressed, allowing transcriptional readthrough at tR′. The PR′ promoter drives transcription beyond tR′, leading to the expression of Shiga toxin genes. In the absence of StxS, rpoS is constitutively expressed.(d, e) Role of FasX and MTS1338 in immune evasion and persistence:Schematic representations of the regulatory roles of FasX and MTS1338 sRNAs in evading the host immune response and promoting bacterial persistence within the host environment.
Figure 1.
Role of Bacterial sRNAs in Virulence (a) Regulation of P-pili expression by PapR: The expression of P-pili in uropathogenic Escherichia coli (UPEC) is controlled by a phase switch that toggles between the OFF and ON states. Upper left panel: In the OFF state, the phase switch is regulated by the interaction of Lrp, papI, papB, and Dam methylase. The intergenic region between papB and papI contains two sets of methylation sites: proximal and distal. Binding of Lrp to the proximal site prevents Dam methylation, leading to methylation of the distal site and suppression of pap operon transcription, thereby preventing P-fimbriae expression. Upper right panel: In the ON state, Lrp, in association with papI, binds to the proximal site, facilitating activation of the phase switch and allowing transcription of the pap operon, leading to P-fimbriae expression. Lower left panel: The small RNA PapR post-transcriptionally represses papI expression by directly interacting with its transcript, preventing ribosome binding. In the absence of papI, Lrp cannot activate the phase switch, thereby repressing P-fimbriae expression. (b) Structural components of P-fimbriae. A schematic representation of the major structural components of P-fimbriae, which facilitate bacterial adherence to host epithelial cells.(c) Role of StxS in Shiga toxin phage regulation:Left panel (Lysogeny): During lysogeny, the late promoters (PR′) of Stx1 and Stx2 phages are constitutively active, transcribing StxSL, which terminates at the intrinsic terminator tR′. The StxSL transcript is processed by RNase E to generate the regulatory small RNA StxS. StxS enhances the expression of rpoS, the stationary-phase sigma factor, while repressing stx1B, encoding the Shiga toxin 1B subunit, thereby reducing Shiga toxin production. Right panel (Lytic induction): During the lytic cycle, the phage-encoded antiterminator Q is expressed, allowing transcriptional readthrough at tR′. The PR′ promoter drives transcription beyond tR′, leading to the expression of Shiga toxin genes. In the absence of StxS, rpoS is constitutively expressed.(d, e) Role of FasX and MTS1338 in immune evasion and persistence:Schematic representations of the regulatory roles of FasX and MTS1338 sRNAs in evading the host immune response and promoting bacterial persistence within the host environment.
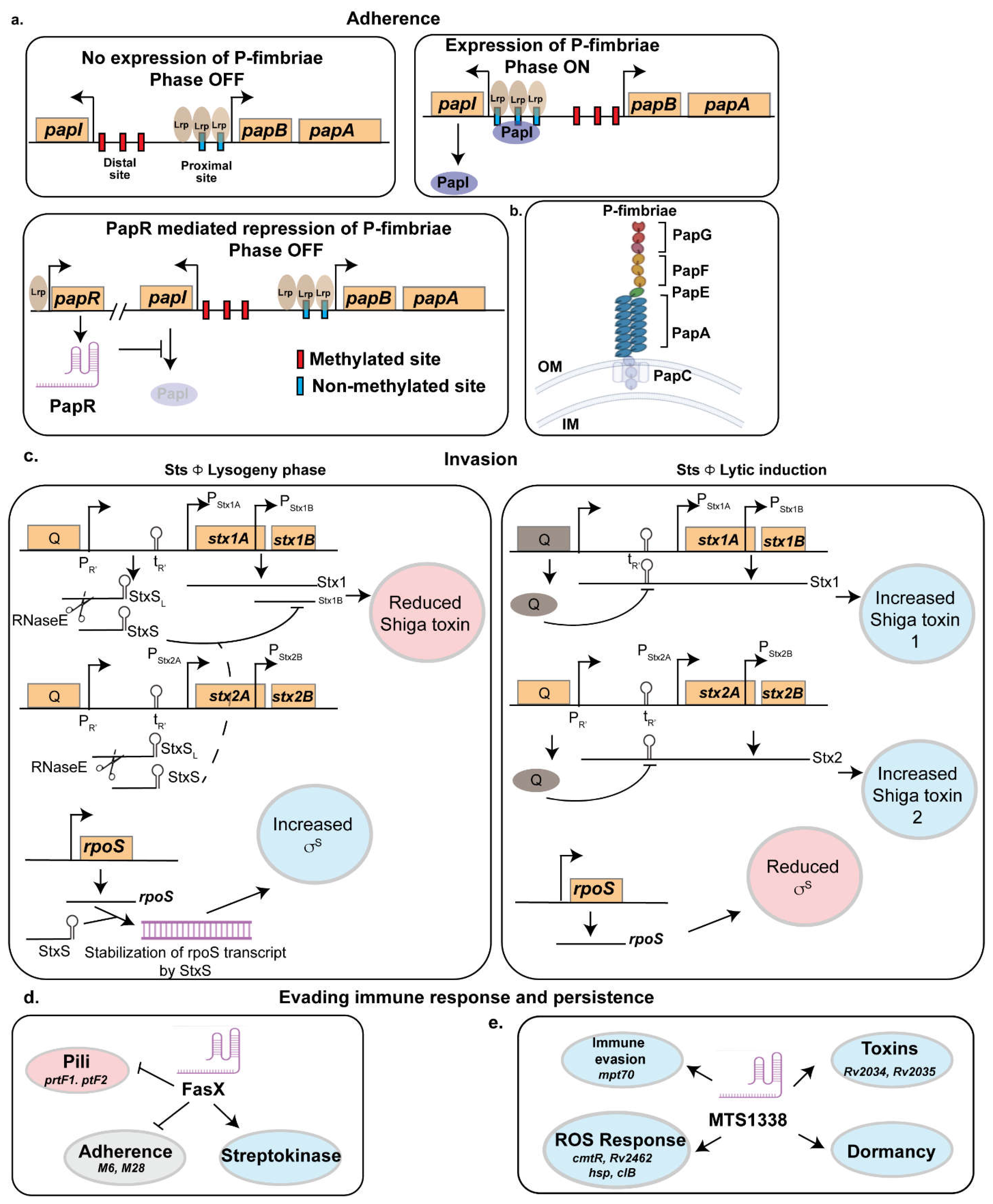
Figure 2.
Role of Small RNAs in Stress Response.(a) Regulation of RyhB under iron-replete conditions:Under Fe-replete condition, the ferric uptake regulator (Fur) is active and represses the transcription of ryhB as well as iron uptake systems, preventing unnecessary iron acquisition.(b) Regulation of RyhB under iron-limiting conditions:Under iron limiting conditions, Fur becomes inactive, leading to the derepression of ryhB expression. RyhB facilitates an iron-sparing response by selectively repressing the translation of non-essential iron-dependent proteins while allowing the synthesis of essential Fe- and Fe-S-cluster-containing proteins. In uropathogenic Escherichia coli (UPEC) strain CFT073, RyhB also contributes to an anticipatory stress response, equipping the pathogen for survival in the nutrient-limited environment of the urinary tract. (c) Role of OxyS in oxidative stress response:OxyS is induced under oxidative stress conditions by the transcriptional regulator OxyR. OxyS modulates downstream gene expression to mediate DNA damage repair and induce cell cycle arrest, thereby preventing oxidative damage.(d) Role of RybB in envelope stress response:RybB is activated by the extracytoplasmic stress sigma factor, σE (SigE), under envelope stress conditions. It promotes the synthesis of outer membrane proteins to facilitate membrane repair while simultaneously repressing biofilm formation, thereby balancing cellular adaptation to stress.
Figure 2.
Role of Small RNAs in Stress Response.(a) Regulation of RyhB under iron-replete conditions:Under Fe-replete condition, the ferric uptake regulator (Fur) is active and represses the transcription of ryhB as well as iron uptake systems, preventing unnecessary iron acquisition.(b) Regulation of RyhB under iron-limiting conditions:Under iron limiting conditions, Fur becomes inactive, leading to the derepression of ryhB expression. RyhB facilitates an iron-sparing response by selectively repressing the translation of non-essential iron-dependent proteins while allowing the synthesis of essential Fe- and Fe-S-cluster-containing proteins. In uropathogenic Escherichia coli (UPEC) strain CFT073, RyhB also contributes to an anticipatory stress response, equipping the pathogen for survival in the nutrient-limited environment of the urinary tract. (c) Role of OxyS in oxidative stress response:OxyS is induced under oxidative stress conditions by the transcriptional regulator OxyR. OxyS modulates downstream gene expression to mediate DNA damage repair and induce cell cycle arrest, thereby preventing oxidative damage.(d) Role of RybB in envelope stress response:RybB is activated by the extracytoplasmic stress sigma factor, σE (SigE), under envelope stress conditions. It promotes the synthesis of outer membrane proteins to facilitate membrane repair while simultaneously repressing biofilm formation, thereby balancing cellular adaptation to stress.
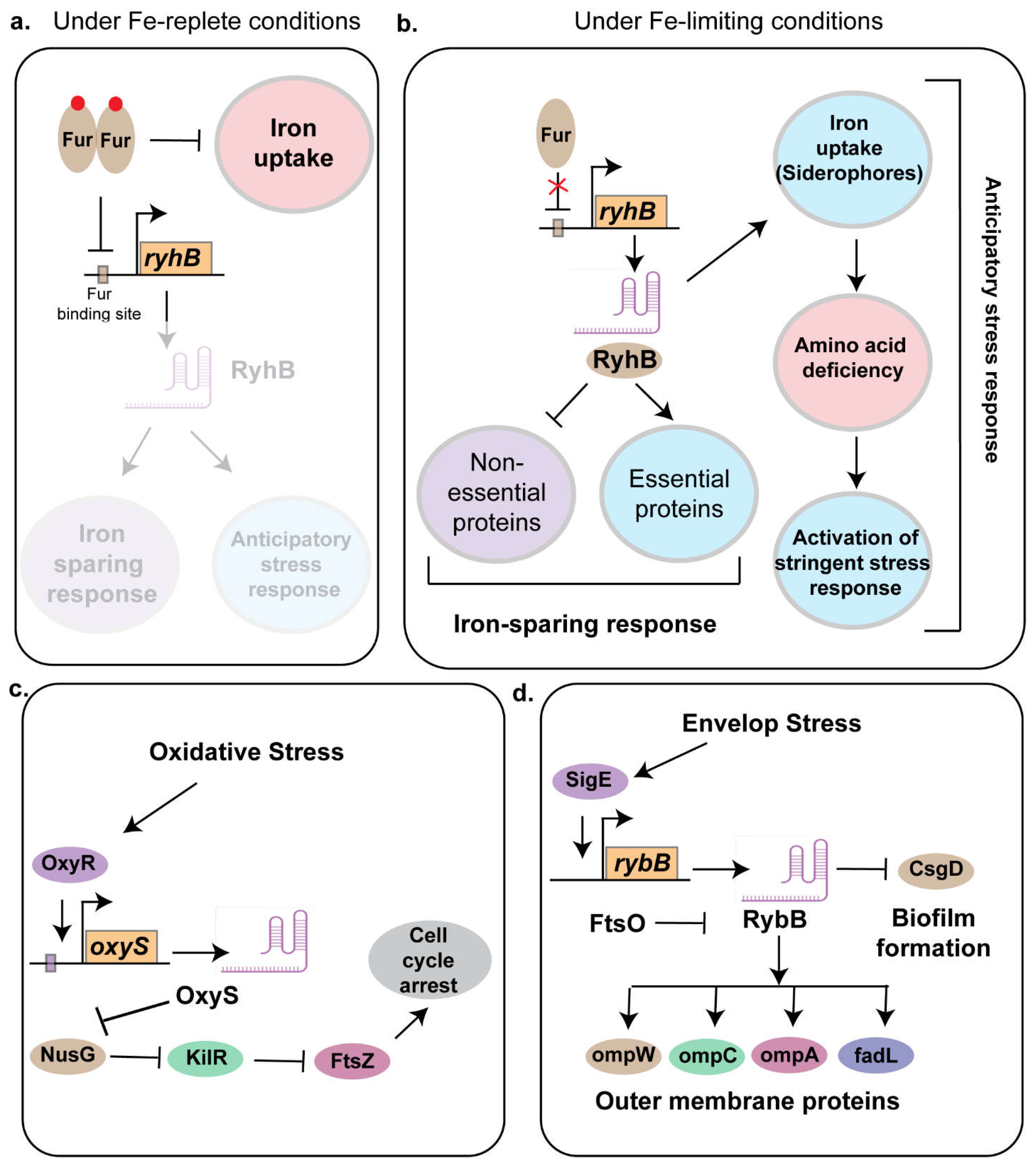
Figure 3.
Mechanisms of Bacterial Antibiotic Resistance(a) Small RNA-mediated antibiotic resistance mechanisms:Upon exposure to environmental stimuli, bacterial pathogens activate transcriptional regulators, which in turn induce the expression of cognate sRNAs. These sRNAs contribute to antibiotic resistance by either upregulating efflux pumps that expel antibiotics from the cell or downregulating outer membrane proteins involved in antibiotic uptake, thereby reducing intracellular antibiotic accumulation.(b, c) Role of small RNAs in antibiotic resistance through biofilm formation:(b) Role of AS1974 in biofilm formation:The sRNA AS1974 is activated through epigenetic regulation and functions to inhibit antibiotic resistance, biofilm formation, and bacterial adherence to the host.(c) Role of PhoS in biofilm formation:Under phosphate-limiting conditions, the sensor kinase PhoR undergoes autophosphorylation and subsequently phosphorylates the response regulator PhoB. Activated PhoB induces the expression of phoS, an sRNA that serves as a positive regulator of biofilm formation. Additionally, PhoS post-transcriptionally stabilizes phoR mRNA, reinforcing a positive feedback loop that sustains the phosphate starvation response.Conversely, under phosphate-replete conditions, PhoR functions as a phosphatase, dephosphorylating PhoB, which in turn prevents phoS expression. The absence of PhoS leads to reduced biofilm formation, as indicated by the masked region in the figure.
Figure 3.
Mechanisms of Bacterial Antibiotic Resistance(a) Small RNA-mediated antibiotic resistance mechanisms:Upon exposure to environmental stimuli, bacterial pathogens activate transcriptional regulators, which in turn induce the expression of cognate sRNAs. These sRNAs contribute to antibiotic resistance by either upregulating efflux pumps that expel antibiotics from the cell or downregulating outer membrane proteins involved in antibiotic uptake, thereby reducing intracellular antibiotic accumulation.(b, c) Role of small RNAs in antibiotic resistance through biofilm formation:(b) Role of AS1974 in biofilm formation:The sRNA AS1974 is activated through epigenetic regulation and functions to inhibit antibiotic resistance, biofilm formation, and bacterial adherence to the host.(c) Role of PhoS in biofilm formation:Under phosphate-limiting conditions, the sensor kinase PhoR undergoes autophosphorylation and subsequently phosphorylates the response regulator PhoB. Activated PhoB induces the expression of phoS, an sRNA that serves as a positive regulator of biofilm formation. Additionally, PhoS post-transcriptionally stabilizes phoR mRNA, reinforcing a positive feedback loop that sustains the phosphate starvation response.Conversely, under phosphate-replete conditions, PhoR functions as a phosphatase, dephosphorylating PhoB, which in turn prevents phoS expression. The absence of PhoS leads to reduced biofilm formation, as indicated by the masked region in the figure.
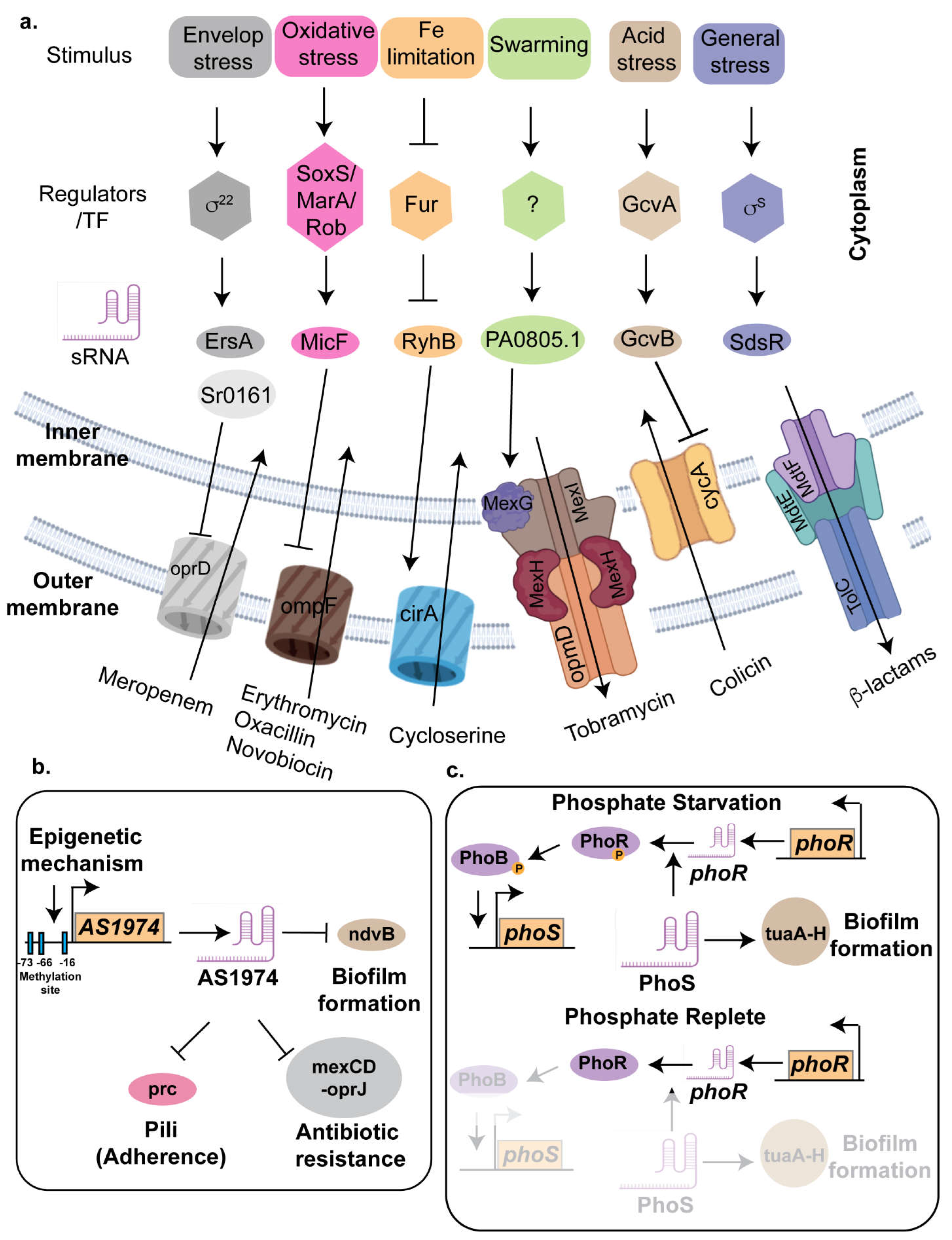
Figure 4.
Role of Qrr sRNAs in Quorum Sensing Regulation in Vibrio spp.(Upper panel: Quorum sensing at low cell density)At low cell density, the concentration of autoinducers is low, leading to the activation of the sensor kinase, which subsequently phosphorylates downstream regulators. This phosphorylation cascade induces the expression of quorum regulatory RNAs (Qrr sRNAs). Qrr sRNAs function to repress the master quorum-sensing regulator LuxR, thereby inhibiting collective behaviors associated with high-density populations. Concurrently, Qrr activates AphA, a key regulator of virulence gene expression, enhancing pathogenicity. Additionally, Qrr sRNAs suppress the Type VI Secretion System (T6SS), a mechanism involved in interbacterial competition.(Lower panel: Quorum sensing at high cell density)At high cell density, the accumulation of autoinducers triggers a shift in sensor kinase activity from phosphorylation to phosphatase activity, leading to the degradation of Qrr sRNAs. In the absence of Qrr, LuxR expression is derepressed, promoting collective behaviors characteristic of quorum sensing, including biofilm formation and luminescence. Additionally, derepression of LuxR enhances T6SS activity while simultaneously repressing virulence gene expression, shifting the bacterial population toward cooperative rather than pathogenic behaviors.
Figure 4.
Role of Qrr sRNAs in Quorum Sensing Regulation in Vibrio spp.(Upper panel: Quorum sensing at low cell density)At low cell density, the concentration of autoinducers is low, leading to the activation of the sensor kinase, which subsequently phosphorylates downstream regulators. This phosphorylation cascade induces the expression of quorum regulatory RNAs (Qrr sRNAs). Qrr sRNAs function to repress the master quorum-sensing regulator LuxR, thereby inhibiting collective behaviors associated with high-density populations. Concurrently, Qrr activates AphA, a key regulator of virulence gene expression, enhancing pathogenicity. Additionally, Qrr sRNAs suppress the Type VI Secretion System (T6SS), a mechanism involved in interbacterial competition.(Lower panel: Quorum sensing at high cell density)At high cell density, the accumulation of autoinducers triggers a shift in sensor kinase activity from phosphorylation to phosphatase activity, leading to the degradation of Qrr sRNAs. In the absence of Qrr, LuxR expression is derepressed, promoting collective behaviors characteristic of quorum sensing, including biofilm formation and luminescence. Additionally, derepression of LuxR enhances T6SS activity while simultaneously repressing virulence gene expression, shifting the bacterial population toward cooperative rather than pathogenic behaviors.
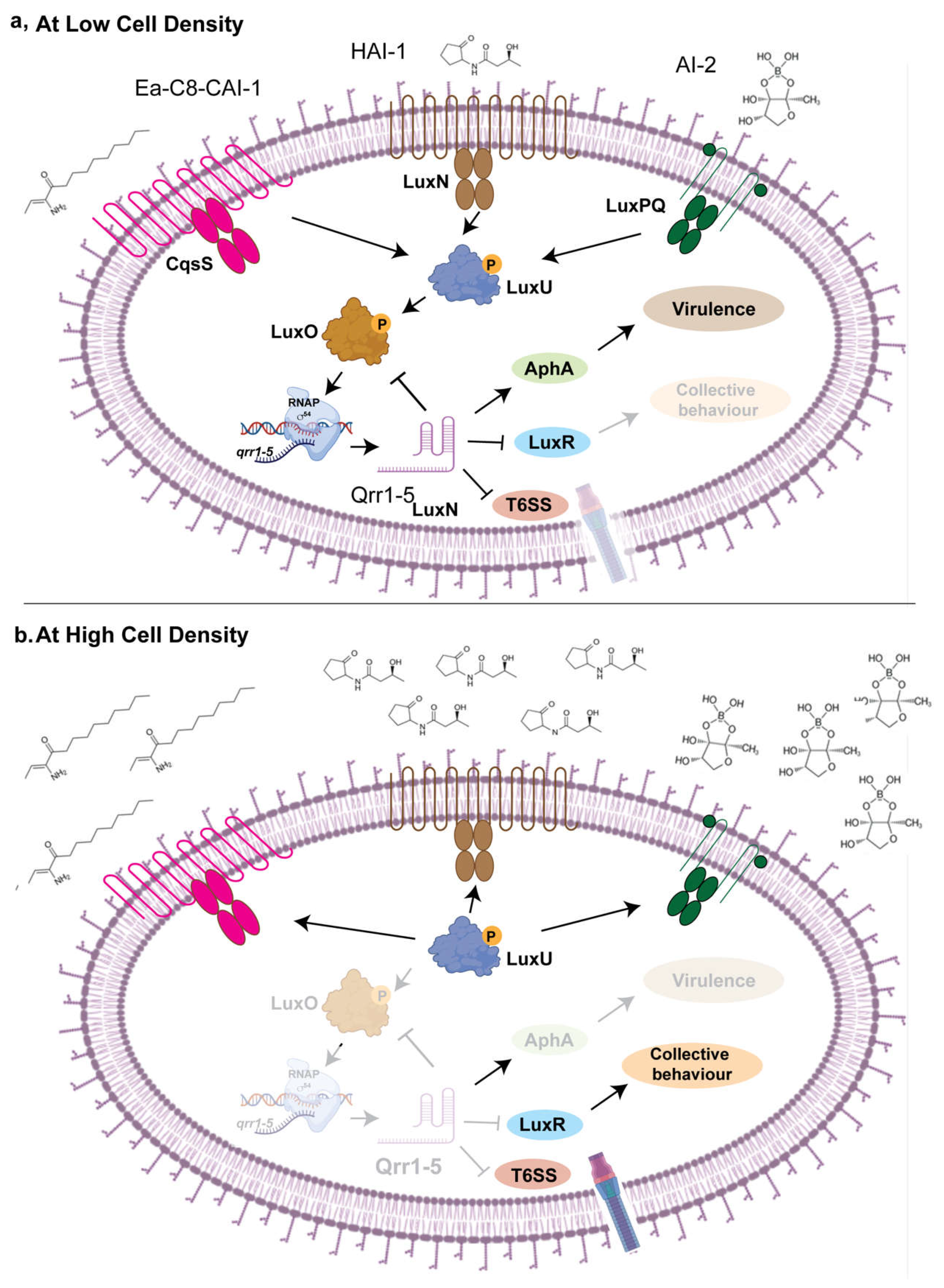
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



