
Submitted:
03 March 2025
Posted:
04 March 2025
You are already at the latest version
Abstract
Troponin C (TnC) is the Ca2+-sensing subunit of troponin that is responsible for activating thin filaments in striated muscle, and in turn for regulating systolic and diastolic contractile function of cardiac muscle. Secondary structure of vertebrate TnC is mainly α-helices, with 9 helices named from N and A-H. The N-helix is a 12-residue-long α-helix located at the extreme amino terminus of the protein and is the only helical structure that does not participate in forming Ca2+-binding EF-hands. Evolutionarily, the N-helix is found only in TnC from mammalian species and most other vertebrates and is not present in other Ca2+-binding protein members of the calmodulin (CaM) family. Furthermore, the primary sequence of the N-helix differs between the genetic isoforms of fast skeletal TnC (sTnC) and cardiac/slow skeletal TnC (cTnC). The 3D location of N-helix within troponin complex is also distinct between skeletal and cardiac troponin. Physical chemistry and biophysical studies centered on the sTnC N-helix demonstrate that it is crucial to thermal stability and Ca2+-sensitivity of thin filament regulated MgATPase activity in solution and isometric force generation in the sarcomere. Comparable studies on cTnC N-helix have not yet been performed despite identification of cardiomyopathy-associated genetic variants that affect residues of cTnC’s N-helix. Here we review the current status of research on TnC’s N-helix and establish future directions to elucidate its functional significance.
Keywords:
troponin C (TnC)
; N-terminal α-helix
; thin filament activation
; cryo-electron microscopy (cryo-EM)
; cardiomyopathy
1. Introduction
1.1. Troponin C Is Essential for Ca2+-Regulation of Striated Muscle Contraction
Calcium ions (Ca2+) play an essential role in muscle contraction [1,2,3,4]. The Ca2+-sensor and final mediator of excitation-contraction coupling of striated muscles is troponin C (TnC), one of three distinct polypeptide subunits that comprise the troponin complex (Tn) component of the thin filaments in striated muscles [5,6,7,8,9,10,11,12,13]. TnC, in concert with the other components of the troponin ternary complex and tropomyosin, controls the ‘on’ and ‘off’ states of the thin filament, and thus actomyosin interactions and force generation, by sensing and responding to cytoplasmic Ca2+ concentration [14,15,16,17,18,19,20,21,22,23,24,25,26]. Associated with its role as a Ca2+-binding protein, the abundance of TnC in striated muscles is sufficiently high, therefore TnC also functions as a major cytoplasmic Ca2+ buffer [27,28,29,30]. Troponin subunits may also have non-canonical roles in other cellular compartments such as the nucleus as well as in other tissues such as the brain. Therefore, elucidation of novel non-canonical roles for thin filament proteins is an emerging topic and has been described elsewhere [31,32,33,34,35,36,37,38,39,40,41,42,43,44].
1.2. TnC Function as a Ca2+-Sensor Is Intimately Related to Its Structure
In vertebrates, there are two genetic isoforms of TnC [45,46] that have important similarities and differences in their structure and function. One TnC isoform is the gene product of TNNC2 that is expressed in fast skeletal muscle (skeletal TnC, sTnC) [47,48]. The other TnC isoform is the gene product of TNNC1 that is expressed in both cardiac and slow skeletal muscles [47,49,50]; here we will focus on its role in cardiac muscle (cardiac TnC, cTnC) (Figure 1). Physical chemistry techniques show that both isoforms of TnC contain significant amounts of α-helical structure and this structure is quite stable at supraphysiological temperatures when divalent cations, especially Ca2+, are present [51,52,53,54]. Bioinformatics and structural analyses show in greater detail that there are nine distinct α-helices organized into a tertiary structure that resembles a dumbbell in shape with an amino terminal lobe (N-lobe) and a carboxyl terminal lobe (C-lobe) that are connected by a linker sequence that is more structured in sTnC than in cTnC [9,55,56,57,58,59,60,61,62] (Figure 1). The first α-helix of TnC, located at the extreme amino terminus, is referred to as the N-helix (Figure 1). When progressing through the primary sequence from the N-helix toward the C-terminus, the other eight α-helices are paired sequentially (helix pairs A-B, C-D, E-F, G-H) to form four EF-hand structures, two in the N-lobe (sites I and II) and two in the C-lobe (sites III and IV) (Figure 2a, b).
1.3. TnC EF-Hand Affinity and Selectivity Determines Function
The physiological relevance of the TnC N-lobe and C-lobe EF-hands has been assessed by examining their affinities and selectivities relative to cytoplasmic Ca2+ and Mg2+ levels. Comparison of TnC EF-hand affinities with physiologically relevant levels of divalent cations indicates that TnC N-lobe regulates striated muscle contraction in a Ca2+-dependent manner while the C-lobe should always be occupied by Ca2+ or Mg2+. Cytoplasmic Ca2+ at rest is on the order of 10-7 M and can rise by an order of magnitude in a cardiomyocyte during systole, and by two orders of magnitude or more during excitation of a vertebrate skeletal muscle fiber. While the peak of the Ca2+ transient is greater in vertebrate skeletal muscle than in cardiac muscle, the duration of skeletal muscle Ca2+ transients is typically much shorter [10,27,63,64,65]. Cytoplasmic Mg2+ is generally in the range of 0.3 – 3 mM; additional Mg2+ is present in the cytoplasm complexed with ATP (MgATP) and also with the EF-hand protein parvalbumin, if present [66,67,68,69].
In both sTnC and cTnC, C-lobe sites III and IV can bind either Mg2+ or Ca2+ with relatively high affinity (order of 10-8 – 10-6 M for Ca2+ binding, depending on Mg2+ concentration due to binding competition, and order of 10-4 for Mg2+ binding). At these affinities both sites III and IV should always be occupied with one of these two divalent cations, including when the muscle is relaxed (diastole in the heart) [70,71,72]. This is essential for the C-lobe to remain bound to TnI under physiological conditions, a critical structural role.
There is general consensus that the N-lobes of sTnC and cTnC, in comparison with the C-lobes, are more selective for Ca2+ over Mg2+ under physiological conditions [1,9,53,58,70,71,72,73,74], although there is one report that suggests Mg2+ may bind weakly to the N-lobe of cTnC [75]. The two EF-hands in the N-lobe of sTnC can each bind one Ca2+ at activating Ca2+ levels. In contrast, the N-lobe of cTnC can only bind Ca2+ at site II because under physiological conditions the sequence of cTnC site I is incompetent for Ca2+ binding [1,58,70,72,76,77] (Figure 1). Thus, the primary function of Ca2+-binding to the N-lobe during a Ca2+-transient is to activate the thin filament, permitting crossbridge cycling and muscle contraction. Interestingly, the N- and C-lobes of TnC can each modulate the function of the other lobe. For example, a cardiomyopathy mutation at site IV in the C-lobe of cTnC not only abolishes divalent cation binding to the C-lobe but has also been reported to alter Ca2+-affinity of the N-lobe [78].
Figure 1.
Cardiac troponin C (cTnC, bottom left) is the myofilament Ca2+ sensor for excitation-contraction coupling in the heart (top left). Clockwise from top left: contractile cells that comprise much of the walls of the heart are cardiomyocytes. Working together in a coordinated manner, cardiomyocytes pump blood from the chambers of the heart into the circulation. Within cardiomyocytes, contractile myofibrils consist of sarcomeres arranged in series. Alignment of sarcomeres results in striations that are perpendicular to the axis of contraction of myofibrils and cardiomyocytes (note that the orientation of structures on the right half of the figure is perpendicular the axis of contraction for cardiomyocytes). Sarcomeres are comprised of highly organized arrays of myosin-containing thick filaments (pink structures in the sarcomere center) interdigitated with actin-tropomyosin-troponin containing thin filaments (linear purple structures on both halves of the sarcomere); giant elastic protein titin (spring-like structure in the sarcomere) is the largest protein expressed from the human genome and each titin molecule extends from one end to the middle of the sarcomere. The detailed structure of the repeating structural unit of the cardiac thin filament determined by cryo-EM is shown below the sarcomere: actin is gray, tropomyosin is green, troponin T is yellow, troponin I is blue, and cTnC is red (PDB: 7K05) [20]. Note that the two strands of tropomyosin-troponin are offset relative to each other due to the helical structure of the actin filament; detailed structural analysis shows that the two strands are not equivalent and are referred to as the upper and lower strand [20,22]. cTnC has a tertiary structure that resembles a dumbbell, with an amino terminal lobe (N-lobe, top) and a carboxyl terminal lobe (C-lobe, bottom) connected by a linker sequence (center red). The N-helix of cTnC (the focus of this review) is highlighted in violet. cTnC structure adapted from the Ca2+-bound state of the upper strand of the native porcine cardiac thin filament determined by cryo-EM (PDB: 8UZX chain C) [22]. Three Ca2+ ions are shown as green spheres with one bound at cTnC regulatory site II in the N-lobe and one bound at each of sites III and IV in the C-lobe.
Figure 1.
Cardiac troponin C (cTnC, bottom left) is the myofilament Ca2+ sensor for excitation-contraction coupling in the heart (top left). Clockwise from top left: contractile cells that comprise much of the walls of the heart are cardiomyocytes. Working together in a coordinated manner, cardiomyocytes pump blood from the chambers of the heart into the circulation. Within cardiomyocytes, contractile myofibrils consist of sarcomeres arranged in series. Alignment of sarcomeres results in striations that are perpendicular to the axis of contraction of myofibrils and cardiomyocytes (note that the orientation of structures on the right half of the figure is perpendicular the axis of contraction for cardiomyocytes). Sarcomeres are comprised of highly organized arrays of myosin-containing thick filaments (pink structures in the sarcomere center) interdigitated with actin-tropomyosin-troponin containing thin filaments (linear purple structures on both halves of the sarcomere); giant elastic protein titin (spring-like structure in the sarcomere) is the largest protein expressed from the human genome and each titin molecule extends from one end to the middle of the sarcomere. The detailed structure of the repeating structural unit of the cardiac thin filament determined by cryo-EM is shown below the sarcomere: actin is gray, tropomyosin is green, troponin T is yellow, troponin I is blue, and cTnC is red (PDB: 7K05) [20]. Note that the two strands of tropomyosin-troponin are offset relative to each other due to the helical structure of the actin filament; detailed structural analysis shows that the two strands are not equivalent and are referred to as the upper and lower strand [20,22]. cTnC has a tertiary structure that resembles a dumbbell, with an amino terminal lobe (N-lobe, top) and a carboxyl terminal lobe (C-lobe, bottom) connected by a linker sequence (center red). The N-helix of cTnC (the focus of this review) is highlighted in violet. cTnC structure adapted from the Ca2+-bound state of the upper strand of the native porcine cardiac thin filament determined by cryo-EM (PDB: 8UZX chain C) [22]. Three Ca2+ ions are shown as green spheres with one bound at cTnC regulatory site II in the N-lobe and one bound at each of sites III and IV in the C-lobe.

Comparable to the EF-hands, the N-helix of TnC is also an important structure involved in thin filament regulation although its distinct function(s) are not quite clear yet. Hence, in this review, we are going to combine currently available evidence from multiple studies to discuss how this helical structure participates in normal function of the thin filament.
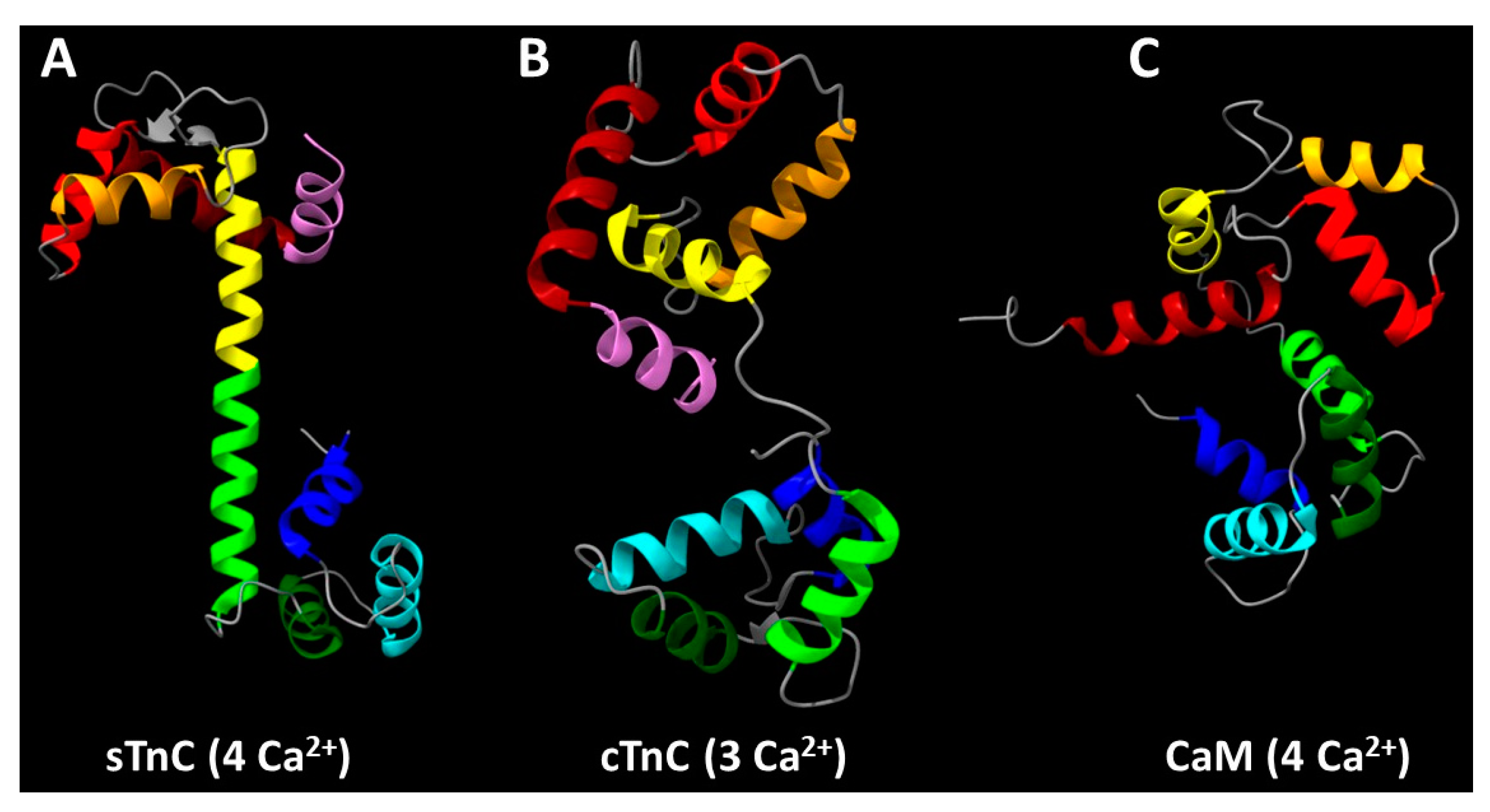
Figure 2.
The arrangement of α-helices in (A) skeletal troponin C (sTnC), (B) cardiac troponin C (cTnC), and (C) calmodulin (CaM). Starting from the amino terminus, sTnC and cTnC have an N-helix (highlighted in violet) that is absent in CaM and other calmodulin family Ca2+-binding proteins. Continuing toward the carboxyl terminus in all three proteins, the sequentially labeled α-helices occur in pairs that form EF-hand motifs: helices A-B (dark red and red, respectively) and C-D (orange and yellow, respectively) are in the N-lobe, and helices E-F (lime green and dark green, respectively) and G-H (cyan and blue, respectively) are in the C-lobe. Structure of sTnC was adapted from the Ca2+-bound state of the core domain of recombinant chicken skeletal troponin determined by X-ray crystallography (PDB: 1YTZ chain C) according to Vinogradova et al. [79]. Structure of cTnC was adapted from the Ca2+-bound state of the upper strand of the native porcine cardiac thin filament determined by cryo-EM (PDB: 7KO5 chain V) according to Risi et al. [20]. Structure of CaM was adapted from the Ca2+-bound state of recombinant Drosophila melanogaster CaM (complexed with a synthetic peptide corresponding to the CaM-binding domain of rabbit skeletal muscle myosin light chain kinase) determined by solution NMR spectroscopy (PDB: 2BBM chain A) according to Ikura et al. [80].
Figure 2.
The arrangement of α-helices in (A) skeletal troponin C (sTnC), (B) cardiac troponin C (cTnC), and (C) calmodulin (CaM). Starting from the amino terminus, sTnC and cTnC have an N-helix (highlighted in violet) that is absent in CaM and other calmodulin family Ca2+-binding proteins. Continuing toward the carboxyl terminus in all three proteins, the sequentially labeled α-helices occur in pairs that form EF-hand motifs: helices A-B (dark red and red, respectively) and C-D (orange and yellow, respectively) are in the N-lobe, and helices E-F (lime green and dark green, respectively) and G-H (cyan and blue, respectively) are in the C-lobe. Structure of sTnC was adapted from the Ca2+-bound state of the core domain of recombinant chicken skeletal troponin determined by X-ray crystallography (PDB: 1YTZ chain C) according to Vinogradova et al. [79]. Structure of cTnC was adapted from the Ca2+-bound state of the upper strand of the native porcine cardiac thin filament determined by cryo-EM (PDB: 7KO5 chain V) according to Risi et al. [20]. Structure of CaM was adapted from the Ca2+-bound state of recombinant Drosophila melanogaster CaM (complexed with a synthetic peptide corresponding to the CaM-binding domain of rabbit skeletal muscle myosin light chain kinase) determined by solution NMR spectroscopy (PDB: 2BBM chain A) according to Ikura et al. [80].
2. Structural and Functional Studies Suggest a Critical Role for the N-Helix of Cardiac Troponin C in Ca2+-Regulation of Cardiac Muscle
2.1. Evolutionary Significance of TnC N-Helix
TnCs, along with myosin light chains, parvalbumins and calmodulin (CaM) itself, are part of the calmodulin family of EF-hand, Ca2+-binding proteins [81]. Interestingly, of the large number of proteins in the calmodulin family, only vertebrate TnCs have sequences that correspond to an N-helix, the one α-helix of TnC that is not directly involved in the formation of an EF-hand [81,82,83]. This can be seen in Figure 2, which illustrates the presence of an N-helix (violet)—adjacent to the A-helix (dark red) and B-helix (red) that surround EF-hand site I—in sTnC (Figure 2a) and cTnC (Figure 2b), while there is no sequence or structure comparable to the N-helix preceding the first helix-loop-helix domain in CaM (Figure 2c).
While the evolutionary origin of the N-helix of TnC is not known, it is unique to vertebrate striated muscles [81,83]. There is diversity among invertebrate TnC’s in regard to which of the four EF-hands is functionally capable of binding divalent cations or presumed to be functional based upon sequence analysis [84,85,86,87]. In stark contrast to vertebrate TnC discussed above, the C-lobe of TnC appears to be more significant than the N-lobe for Ca2+-activation of most invertebrate striated muscles [11,84,85,87]. Thus, it seems likely that the N-helix could have a significant role in Ca2+-activation of vertebrate striated muscle, although the exact nature of that role could differ between cardiac and skeletal TnC. The α-helical structures at the N-termini of sTnC and cTnC appear similar (Figure 2a,b), but the sequences differ among species, particularly for sTnC, and isoforms within a species [47,48,49,81,83]. The latter point is illustrated in Figure 3 for the human genetic isoforms. Among the first 12 amino acids, only four are identical (Figure 3) and one of those four is the N-terminal Met that is typically removed post-translationally from sTnC but not cTnC of vertebrate species [48,49,76,81,88,89,90,91,92]. Protein sequencing indicates that the N-terminus of both cTnC and sTnC is further modified post-translationally by acetylation of the remaining N-terminal amino group [48,49,76,88,89,90,91] which neutralizes what would otherwise be the positive charge of the N-terminus of the mature polypeptide. These differences in primary sequence between the N-helices of sTnC and cTnC are substantial enough to suggest that they may not be functionally identical. Thus, the exact nature of the role of the N-helix in Ca2+-activation of vertebrate striated muscle could differ between cardiac and skeletal TnC.
2.2. Structural Evidence for the Functional Significance of the TnC N-Helix
High resolution structural studies have provided important insights into Ca2+-activation of the thin filament by applying an evolving variety of structural determination methods to elucidate the structure of TnC. The structures include isolated TnC, TnC associated with portions of or all of the troponin complex, and TnC within the thin filament, in the absence and presence of divalent cations in the N-lobe site (cTnC) or sites (sTnC).
X-ray crystallography provided the earliest structures of native sTnC purified from chicken or turkey skeletal muscles [55,56,57]. These studies supplied structural details not available in an earlier report of diffraction from crystals of rabbit and chicken sTnC [93]. All of the earliest crystal structures had two metal ions bound to the C-lobe, with 2 Ca2+ in the turkey sTnC structures [55,56] and either 2 Mn2+, 2 Nd3+ or 2 Au3+ in the chicken sTnC structures [57,93]. In these structures, the apo N-lobe and metal ion-bound C-lobe take on similar but distinct conformations that allowed for predictions about the structural changes that occur upon regulatory Ca2+ binding in the N-lobe. Almost a decade later, Houdusse et al. [60] filled in the gap by obtaining crystal structures of recombinantly expressed chicken sTnC with 4 Ca2+ bound, thereby demonstrating experimentally what could only be speculated upon in earlier studies. None of these studies provided a structure with physiological Mg2+ bound at the C-lobe.
The D/E-linker is of particular note among all of these sTnC structures because it forms a single, extended α-helix separating the N- and C-lobes. When analogizing sTnC structure to a dumbbell, the central D/E-linker forms the handlebar between the N- and C-lobe masses at either end (Figure 2a) [55,56,57,60]. The linear D/E-linker separating the N- and C-lobes in all of these sTnC structures could have resulted from the conditions of crystallization (e.g., acidic pH ~ 5) and structural constraints within the crystal lattices formed. In addition, the structures may have been affected by the absence of the other troponin complex proteins, especially the portions of TnI that bind the N- and C-lobes when divalent cations are present [9,94,95,96,97,98]. An X-ray crystallography study by Saijo et al. [99] demonstrated flexibility of the D/E-linker of recombinant rabbit sTnC in the 2 Ca2+ state (i.e., apo N-lobe) crystallized at basic pH (pH ~ 8) in the presence of sTnI1-47 that binds the C-lobe. Solution NMR spectroscopy further demonstrated flexibility of the D/E-linker of Ca2+-saturated recombinant chicken sTnC (4 Ca2+) [59] and recombinant Cys-less chicken cTnC (3 Ca2+) [61]. In solution, i.e., without the constraints of a crystal lattice, the N- and C-lobe motions are essentially independent—recognizing that they are not completely independent because they are covalently linked to each other—and thus for many purposes the N- and C-lobes can be studied as separate constructs [94,95,96,100,101,102]. While the structures discussed above included the N-helix, a major focus was on divalent cation-induced structural changes within the N- and C-lobes, and little attention was paid to the N-helix. It was even concluded from solution NMR studies that the absence of the N-helix had little or no effect on the proteolytically isolated, apo N-lobe of native turkey sTnC [100,103]. A similar conclusion was obtained from lower resolution circular dichroism studies on recombinant chicken sTnC [54], although the thermal stability of chicken and rabbit sTnC structure was reduced in the absence of the N-helix regardless of divalent cation binding [51,54].
Building upon the work of Saijo et al. [99], Takeda et al. [62] determined the X-ray crystal structure of Ca2+-saturated (3 Ca2+) cardiac troponin core domain. They showed a collapsed—albeit not fully resolved—DE-linker. In the Takeda et al. [62] structure, the N-helix of cTnC’s N-lobe is in close proximity to the C-lobe and, more specifically, the portion of cTnI that is bound to the C-lobe. Within two years of the Takeda et al. publication [62], structures became available for the skeletal troponin core domain in both the Ca2+-free, Mg2+-saturated state (apo N-lobe and 2 Mg2+ C-lobe) as well as the Ca2+-saturated (4 Ca2+) state [79]. In contrast to the Ca2+-saturated cardiac troponin core domain of Takeda et al. [62], the Ca2+-saturated (4 Ca2+) skeletal troponin core domain exhibited an elongated, α-helical D/E-linker [79] comparable to that observed in the crystal structures of sTnC with only 2 Ca2+ bound to the C-lobe [55,56,57,60]. However, the D/E-linker of the skeletal troponin core domain becomes disordered in the Ca2+-free (2 Mg2+) state. Ca2+-saturated TnC from these crystal structures of the skeletal and cardiac troponin complexes are shown in Figure 2a,b, respectively, illustrating what was inferred from sequence differences between the N-helices of sTnC and cTnC—that the role of the N-helix in Ca2+-activation of vertebrate striated muscle could differ between cTnC and sTnC.
With the advancement of cryo-EM—now the ‘gold-standard’ technology of macromolecular structure analysis—we have been able to visualize structural changes within the thin filament upon Ca2+ binding to cTnC, and to more deeply analyze its regulatory mechanism [19,20,22,104,105]. While an individual structural model determined by cryo-EM represents a single structural state, capture of multiple structural states allows cryo-EM to be an excellent technique for the study of thin filament structural dynamics.
Current cryo-EM-based models of the thin filament place the N-helix of cTnC close to three structural components of cTn: cTnC D-helix associated with regulatory Ca2+-binding at EF-hand site II; the D/E-linker connecting the N- and C-lobes of cTnC; and cTnI helix H1 (residues 42-80), located in the N-terminal portion of cTnI that binds to the C-lobe of cTnC, which anchors cTnC within cTn (Figure 1, Figure 4 & Supplemental movies) [19,20,22]. This central location and the altered interactions with other portions of the troponin complex upon Ca2+ binding strongly imply that cTnC N-helix could play a critical role in Ca2+-regulation of cardiac contraction by widely transmitting information about regulatory Ca2+-binding at the cTnC N-lobe to other parts of the cardiac thin filament.
2.3. Biophysical Studies Demonstrate a Critical Role for the N-Helix of sTnC in Normal Ca2+-Regulation of Skeletal Muscle, But Evidence for That of cTnC Is Lacking
Functional assays have been employed to address the question of the functional role(s) of TnC’s N-helix in light of the evidence presented above that (i) evolution has uniquely favored this structural element in vertebrate TnC’s among calmodulin family proteins [60,76,81,82,83,88] and (ii) that the N-helix is situated between TnC’s N-lobe and other parts of troponin on the thin filament [19,20,22,104], while in contrast (iii) that removal of the N-helix has little effect on the secondary structure of TnC [54,100,103]. This research involving sTnC from multiple species has shown that the N-helix does play an essential role in both structure and function of sTnC [51,52,53,54,106,107].
Functionally, the absence of sTnC’s N-helix markedly decreases the Ca2+-affinity of TnC’s regulatory N-lobe. Solution studies have shown that the absence of the N-helix caused a 3-fold change in Kd for Ca2+-binding at chicken sTnC N-lobe—with or without an F29W substitution that provides a fluorescence readout of Ca2+-specific changes in structure [108]—with little or no change in higher-affinity binding of Ca2+ or Mg2+ at the C-lobe [53,54]. The absence of sTnC’s N-helix in reconstituted thin filaments not only reduced the apparent Ca2+-sensitivity by more than 2-fold for actomyosin solution MgATPase activity, but also caused a marked reduction in the maximum activity [53,54]. Interestingly, removal of only part (~½) of the N-helix had little effect on Ca2+-binding to sTnC in solution, but was still associated with a reduction in Ca2+-sensitivity of actomyosin MgATPase activity [52]. Reversing the scenario to examine a non-muscle system that normally relies on CaM, either sTnC or sTnC that is missing the N-helix can replace CaM to activate erythrocyte Ca2+-ATPase, albeit with a requirement for substantially higher Ca2+ levels [109]; in this non-muscle system, less Ca2+ is required with sTnC that is missing the N-helix compared to sTnC—the opposite of what is observed in muscle—and markedly higher concentrations of protein are required for either TnC relative to CaM.
A study followed up on the observation that CaM can functionally replace sTnC to regulate isometric force generation by skeletal muscle [110] even though it does not integrate fully into the troponin complex. The fact that CaM does not serve well as a structural replacement of TnC in the troponin complex suggests that one possible role of the N-helix of TnC is to maintain TnC bound to the thin filament. The same group also utilized a recombinant rabbit sTnC construct with the N-helix missing [51,107] to test its impact on skeletal muscle force and found that the highest Ca2+-activated force of skeletal muscle was reduced when the N-helix was missing [51,107]. This effect was similar to CaM, which also reduced Ca2+-sensitivity of steady-state isometric force [110]. However, the authors did not attribute these CaM-associated differences exclusively to the missing N-helix (Figure 2), but instead to incomplete occupancy of thin filament regulatory units by CaM. Interestingly, invertebrate striated muscle where the N-helix of TnC is absent also has reduced Ca2+-sensitivity compared to vertebrate muscle [84].
Removal of chicken sTnC N-helix was associated with a marked reduction in Ca2+-sensitivity of steady-state isometric force generation using recombinantly expressed proteins reconstituted into rabbit skeletal fibers [53,106]. The deletion of the N-helix in these studies was associated with a small reduction in maximum Ca2+-activated force; the difference with studies described above may be due to higher Ca2+ concentrations when studying the N-helix deletion [53,106]. N-helix removal had little or no effect on cooperativity of steady-state Ca2+-binding, MgATPase activity, or isometric force generation, as assessed by the Hill coefficient [53,54,106,111]. Beyond steady-state phenomena, the Ca2+-dependence of the kinetics of isometric tension redevelopment (kTR) were shifted rightward to a similar extent as steady-state isometric force such that there was little effect on the relationship between force and kTR [106].
Based on the discussion above, the N-helix plays a significant role in maintaining the structural stability of TnC and the Ca2+-regulated contraction in skeletal muscle. However, comparable studies have not yet been performed with cTnC. Although the N-helix is evolutionarily favored by vertebrate striated muscle, it is notable (per sequence analysis described in Sec. 2.1 above) that the sequence of cTnC N-helix is not the same as that for sTnC; and also (per structure analysis described in Sec. 2.2 above, esp. Figure 4) that the sTnC N-helix is not tucked into a central location like that of cTnC. Additionally, evidence also supports the structural studies indicating that the N-helix is close to the C-lobe at Ca2+-saturated state in the tertiary structure of cTnC (Supplemental movies) [19,20,61,62], which is not true for sTnC [79].
Secondly, previous research also shows that the second half of N-helix is sufficient to maintain normal sTnC function [52], but this is not true for the cardiac isoform. Multiple amino acid variants spanning the whole helix region are reported to be related to all three major types of cardiomyopathy [112,113,114,115], which will be further discussed in the following section. Thus, it is unreasonable to directly presume that the N-helix of the two TnC isoforms are functionally identical.
2.4. Pathophysiological Evidence for the Significance of the TnC N-Helix: Variants in the N-Helix of Troponin C Are Associated with Human Cardiomyopathies
Although the cTnC N-helix only accounts for 12 amino acids out of 161 residues in total, multiple mutations in this region have been linked to three major types of human cardiomyopathy: dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM) and restrictive cardiomyopathy (RCM) (Figure 5), indicating the importance of the N-helix in regulation of cardiac contraction [112,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138]. For these reasons, attention should be directed towards understanding how the cTnC N-helix participates in the physiological function of the cardiac thin filament and the genesis of cardiomyopathy.
Among the seven mutations identified in the N-helix of cTnC shown in Figure 5, four are published in peer-reviewed journals, and the A8V variant is the most studied. Clinical evidence has shown that the variant relates to both HCM and RCM in human, with different inheritance patterns: autosomal dominant for HCM, while autosomal recessive for RCM [116,117,139,140]. The two-copy variant usually results in an earlier disease onset and poorer prognosis compared with the single-copy variant [140], which is consistent with the data obtained from the murine model [121]. Mechanistic studies examining the A8V variant of cTnC N-helix reveal that this mutation may strengthen the binding of the cTnI switch helix to cTnC, which enhances the Ca2+ affinity of myofibrils without affecting that of isolated cTnC [121,123]. A subsequent study that focused on the same variant showed that the N-helix also appears to communicate with the D-helix of cTnC (part of activating site II), increasing the Ca2+ sensitivity of isometric force generation and modulating both the number of and rate of cycling crossbridges [128]. These mechanical effects are consistent with the A8V variant’s association with HCM, and the structural interpretation is supported by cryo-EM structures shown in Figure 1. Interestingly, another study reported the presence of a sexually dimorphic transcriptome of the A8V variant suggesting a possible novel role of the N-helix in promoting the disease phenotype [130].
Additional evidence suggests that the N-terminal helix of cTnC is close in tertiary structure to the intrinsically disordered C-terminus of cTnT—a structural element that remains unresolved in existing Ca2+-bound cryo-EM structures. This interaction is strengthened in the presence of the I4M pathogenic variant of the cTnC N helix, a variant that is associated with pediatric DCM, probably by negatively influencing the allosteric regulation of Ca2+ activation through the cTnC-cTnI switching mechanism [129].
2.5. Cardiomyopathy Variants May Alter Communication Between the cTnC N-Helix and Other Parts of Troponin
In the cardiac isoform of vertebrate TnC, there are only three divalent cation binding sites, with two sites (site III and site IV) in the C-lobe always occupied and participating in the critical structural role of the C-lobe—binding TnC to TnI—under physiological conditions. In contrast, the remaining site (site II) in the N-lobe responsive to variations in cytoplasmic Ca2+ concentration (Figure 1). The D/E linker is an obvious pathway for communication between the N- and C-lobes of cTnC. Measurements of protein dynamics in solution have shown that the HCM-related cTnC variant (D145E) could enhance the Ca2+ affinity of site II in N-lobe, probably by abnormal hydrophobic surface exposure, although the Ca2+-binding sites in the C-lobe are destabilized [78,141,142].
The cryo-EM structural evidence described above points to the N-helix as an additional pathway. As shown in Figure 4 and Supplemental movies, the N-helix is located at the center of the cTnC D-helix, D/E-linker and cTnI helix H1 in both Ca2+-free and Ca2+-bound states, indicating the possibility of the N-helix as a ‘communicator’ not only between lobes of cTnC, but also subunits of troponin. Thus, this location of the N-helix is likely important for the mechanism by which Ca2+-initiated activation is transmitted from site II in the N-lobe of cTnC to other parts of the thin filament. This presumption is further supported by the pathophysiological research on the A8V variant, which is associated with both HCM and RCM (see Sec. 2.4). Furthermore, the N-helix of cTnC is also presumed to be close to the intrinsically disordered C-terminus of cTnT [129].
3. Conclusions
In summary, we discussed the role of TnC N-helix from evolutionary, structural, physical chemistry, biophysical and pathophysiological perspectives. The N-helix is uniquely found in TnC’s from mammals and most other vertebrates, but not in other members of the calmodulin family of Ca2+-binding proteins, although the N-helix sequence is different in the two isoforms of TnC, fast skeletal TnC (sTnC) and cardiac/slow skeletal TnC (cTnC). Structural comparison of N-helix between the two isoforms also shows two major points: (i) the N-helix of cTnC is located between D-helix, D/E-linker and cTnI helix H1, which is not true for that of sTnC; and (ii) the N-helix of sTnC is closer to the N-lobe at Ca2+-saturated state, while that of the cardiac isoform turned towards C-lobe when Ca2+ binds. Physical chemistry and biophysical studies have shown that N-helix of sTnC is an essential component for determining Ca2+ sensitivity, MgATPase activity and thermal stability, but comparable evidence about cTnC N-helix is not available yet. Nevertheless, studies on pathophysiological cardiomyopathy-relevant variants within the N-helix of cTnC provide evidence that it must have functional significance that parallels or exceeds that of the N-helix in sTnC. Taken together, N-helix of TnC is a vital element of regulation of thin filament activation by Ca2+ in striated muscles, although the N-helix may have distinct functions in the two isoforms of TnC.
4. Future Directions
Until now, functional studies of TnC N-helix have mainly focused on in vitro studies with the sTnC, and thus the cardiac isoform deserves attention to better understand its role in promoting normal cardiac function and in development of cardiomyopathic disease. For this purpose, one remaining issue that needs to be addressed is understanding how the cTnC N-helix dynamically interacts with other parts of troponin and the thin filament during contraction (systolic ejection phase of the cardiac cycle) and relaxation (diastolic filling of the heart). Given the significance of the cTnC N-helix, we hypothesize that a similarly sized polypeptide at the C-terminus of cTnT might also have structural and functional significance even though it has not yet been visualized by cryo-EM. The current, high-resolution structures that have been obtained by cryo-EM are suitable for molecular dynamics (MD) simulations that could help resolve disparities between cryo-EM structures of the cardiac thin filament [142,143,144,145,146]. To this end, fluorescence polarization measurements, some of which were obtained using bifunctional fluorescence probes covalently bonded to residues in the cTnC N-helix may help elucidate this question [147,148]. In addition to in silico simulations, in vitro experiments with cTnC and cardiac muscle proteins are also necessary. Furthermore, it would be beneficial to have one or more animal models that can provide insight into the role of the cTnC N-helix within a physiological context.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Video S1: Movement of cTnC N-helix (magenta) upon Ca2+-binding to cTnC within the troponin complex on the cardiac thin filament; Video S2: Rotated view of movement of cTnC N-helix (magenta) upon Ca2+-binding to cTnC within the troponin complex on the cardiac thin filament. The videos show the dynamic rotation of the cTnC N-helix from Ca2+-free state (PDB 8UWW) to the fully activated state (PDB 8UZX); the two videos show the same Ca2+-dependent movements viewed from perspectives that are related by 90-degree rotation around the F-actin helical axis. The color scheme in the videos is the same as in Figure 4: actin is gray; tropomyosin is green; cTnI is blue; cTnT is yellow; and the cTnC subunit is red with the N-helix sequence highlighted in magenta.
Author Contributions
Conceptualization, Y.S. and P.B.C.; writing—original draft preparation, Y.S., L.A.B, R.K.S and B.M.C; writing—review and editing, C.M.R, M.S.P, J.R.P, V.E.G and P.B.C; visualization, Y.S., L.A.B, R.K.S, B.M.C, C.M.R and V.E.G; supervision, P.B.C; project administration, P.B.C; funding acquisition, J.R.P, V.E.G and P.B.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by U.S. National Institutes of Health NHLBI grant number R01 HL160966 to JRP, VEG and PBC.
Data Availability Statement
No new data were generated for this article. All structure models used in this review are from PDB with the IDs labelled correspondingly in the text.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of Contraction in Striated Muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Rall, J.A. Discovery of the regulatory role of calcium ion in muscle contraction and relaxation: Setsuro Ebashi and the international emergence of Japanese muscle research. Adv. Physiol. Educ. 2022, 46, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Brunello, E.; Fusi, L. Regulating Striated Muscle Contraction: Through Thick and Thin. Annu. Rev. Physiol. 2024, 86, 255–275. [Google Scholar] [CrossRef]
- Ebashi, S.; Endo, M. , Calcium ion and muscle contraction. Prog Biophys Mol Biol 1968, 18, 123–183. [Google Scholar] [PubMed]
- Potter, J. D. , The content of troponin, tropomyosin, actin, and myosin in rabbit skeletal muscle myofibrils. Arch. Biochem. Biophys. 1974, 162, 436–441. [Google Scholar]
- Parmacek, M.S.; Leiden, J.M. Structure, function, and regulation of troponin C. Circulation 1991, 84, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Solaro, R. J.; Rarick, H. M. , Troponin and tropomyosin: proteins that switch on and tune in the activity of cardiac myofilaments. Circ. Res. 1998, 83, 471–80. [Google Scholar]
- Gomes, A.V.; Potter, J.D.; Szczesna-Cordary, D. The Role of Troponins in Muscle Contraction. IUBMB Life 2002, 54, 323–333. [Google Scholar] [CrossRef]
- Li, M.X.; Hwang, P.M. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 2015, 571, 153–166. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Cao, T.; Thongam, U.; Jin, J.-P. Invertebrate troponin: Insights into the evolution and regulation of striated muscle contraction. Arch. Biochem. Biophys. 2019, 666, 40–45. [Google Scholar] [CrossRef]
- Joyce, W.; Ripley, D.M.; Gillis, T.; Black, A.C.; A Shiels, H.; Hoffmann, F.G. A Revised Perspective on the Evolution of Troponin I and Troponin T Gene Families in Vertebrates. Genome Biol. Evol. 2022, 15. [Google Scholar] [CrossRef]
- Russell, B.; Solís, C. Mechanosignaling pathways alter muscle structure and function by post-translational modification of existing sarcomeric proteins to optimize energy usage. J. Muscle Res. Cell Motil. 2021, 42, 367–380. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.; Geeves, M. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Araujo, A.; Walker, J. Phosphate release and force generation in cardiac myocytes investigated with caged phosphate and caged calcium. Biophys. J. 1996, 70, 2316–2326. [Google Scholar] [CrossRef]
- Sun, Y.-B.; Brandmeier, B.; Irving, M. Structural changes in troponin in response to Ca 2+ and myosin binding to thin filaments during activation of skeletal muscle. Proc. Natl. Acad. Sci. 2006, 103, 17771–17776. [Google Scholar] [CrossRef]
- Sun, Y.; Lou, F.; Irving, M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. J. Physiol. 2009, 587, 155–163. [Google Scholar] [CrossRef]
- Fusi, L.; Brunello, E.; Sevrieva, I.R.; Sun, Y.-B.; Irving, M. Structural dynamics of troponin during activation of skeletal muscle. Proc. Natl. Acad. Sci. 2014, 111, 4626–4631. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Risi, C.M.; Pepper, I.; Belknap, B.; Landim-Vieira, M.; White, H.D.; Dryden, K.; Pinto, J.R.; Chase, P.B.; Galkin, V.E. The structure of the native cardiac thin filament at systolic Ca 2+ levels. Proc. Natl. Acad. Sci. 2021, 118. [Google Scholar] [CrossRef]
- Brunello, E.; Marcucci, L.; Irving, M.; Fusi, L. Activation of skeletal muscle is controlled by a dual-filament mechano-sensing mechanism. Proc. Natl. Acad. Sci. 2023, 120. [Google Scholar] [CrossRef]
- Risi, C.M.; Belknap, B.; Atherton, J.; Coscarella, I.L.; White, H.D.; Chase, P.B.; Pinto, J.R.; Galkin, V.E. Troponin Structural Dynamics in the Native Cardiac Thin Filament Revealed by Cryo Electron Microscopy. J. Mol. Biol. 2024, 436, 168498–168498. [Google Scholar] [CrossRef]
- Davis, J.P.; Tikunova, S.B.; Janssen, P.M. L. Mechanisms of Muscle Contraction and Relaxation. In Muscle and Exercise Physiology, Zoladz, J. A., Ed. Academic Press: London, 2019; pp 39-50.
- Kreutziger, K.L.; Gillis, T.E.; Davis, J.P.; Tikunova, S.B.; Regnier, M. Influence of enhanced troponin C Ca2+-binding affinity on cooperative thin filament activation in rabbit skeletal muscle. J. Physiol. 2007, 583, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Tobacman, L.S. Troponin Revealed: Uncovering the Structure of the Thin Filament On-Off Switch in Striated Muscle. Biophys. J. 2020, 120, 1–9. [Google Scholar] [CrossRef]
- Davis, J.P.; Tikunova, S.B. Ca2+ exchange with troponin C and cardiac muscle dynamics. Cardiovasc. Res. 2007, 77, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.; Neher, E.; Taschenberger, H.; Smith, G. Physiology of intracellular calcium buffering. Physiol. Rev. 2023, 103, 2767–2845. [Google Scholar] [CrossRef]
- Bers, D.M.; Zhu, Y. Excitation-Contraction Coupling and Cardiac Contractile Force. J. Cardiovasc. Dis. Res. 2010, 1, 45. [Google Scholar] [CrossRef]
- Smith, G.L.; Eisner, D.A. Calcium Buffering in the Heart in Health and Disease. Circulation 2019, 139, 2358–2371. [Google Scholar] [CrossRef]
- Fakuade, F.E.; Hubricht, D.; Möller, V.; Sobitov, I.; Liutkute, A.; Döring, Y.; Seibertz, F.; Gerloff, M.; Pronto, J.R.D.; Haghighi, F.; et al. Impaired Intracellular Calcium Buffering Contributes to the Arrhythmogenic Substrate in Atrial Myocytes From Patients With Atrial Fibrillation. Circulation 2024, 150, 544–559. [Google Scholar] [CrossRef]
- Fine, R.; Lehman, W.; Head, J.; Blitz, A. , Troponin C in brain. Nature 1975, 258, 260–7. [Google Scholar] [CrossRef]
- Reddy, K.K.; Oitomen, F.M.; Patel, G.P.; Bag, J. Perinuclear localization of slow troponin C m RNA in muscle cells is controlled by a cis-element located at its 3′ untranslated region. RNA 2005, 11, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Alkass, K.; Druid, H.; Bernard, S.; Frisén, J. Identification of cardiomyocyte nuclei and assessment of ploidy for the analysis of cell turnover. Exp. Cell Res. 2011, 317, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Asumda, F.Z.; Chase, P.B. Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation 2012, 83, 106–115. [Google Scholar] [CrossRef]
- Chase, P.B.; Szczypinski, M.P.; Soto, E.P. Nuclear tropomyosin and troponin in striated muscle: new roles in a new locale? J. Muscle Res. Cell Motil. 2013, 34, 275–284. [Google Scholar] [CrossRef]
- Zhang, T.; Taylor, J.; Jiang, Y.; Pereyra, A.S.; Messi, M.L.; Wang, Z.-M.; Hereñú, C.; Delbono, O. Troponin T3 regulates nuclear localization of the calcium channel Cavβ1a subunit in skeletal muscle. Exp. Cell Res. 2015, 336, 276–286. [Google Scholar] [CrossRef]
- Lopez, Y.O.N.; Messi, M.L.; Pratley, R.E.; Zhang, T.; Delbono, O. Troponin T3 associates with DNA consensus sequence that overlaps with p53 binding motifs. Exp. Gerontol. 2018, 108, 35–40. [Google Scholar] [CrossRef]
- Johnston, J.R.; Chase, P.B.; Pinto, J.R. Troponin through the looking-glass: emerging roles beyond regulation of striated muscle contraction. Oncotarget 2017, 9, 1461–1482. [Google Scholar] [CrossRef]
- Kharitonov, A.V.; Shubina, M.Y.; Nosov, G.A.; Mamontova, A.V.; Arifulin, E.A.; Lisitsyna, O.M.; Nalobin, D.S.; Musinova, Y.R.; Sheval, E.V. Switching of cardiac troponin I between nuclear and cytoplasmic localization during muscle differentiation. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2020, 1867, 118601. [Google Scholar] [CrossRef]
- Solís, C.; Solaro, R.J. Novel insights into sarcomere regulatory systems control of cardiac thin filament activation. J. Gen. Physiol. 2021, 153. [Google Scholar] [CrossRef]
- Zhang, T.; Birbrair, A.; Delbono, O. Nonmyofilament-associated troponin T3 nuclear and nucleolar localization sequence and leucine zipper domain mediate muscle cell apoptosis. Cytoskeleton 2013, 70, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Elezaby, A.; Lin, A.J.; Vijayan, V.; Pokhrel, S.; Kraemer, B.R.; Bechara, L.R.G.; Larus, I.; Sun, J.; Baena, V.; Syed, Z.A.; et al. Cardiac troponin I directly binds and inhibits mitochondrial ATP synthase with a noncanonical role in the post-ischemic heart. Nat. Cardiovasc. Res. 2024, 3, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lee, J.; Vincent, L.G.; Wang, Q.; Gu, M.; Lan, F.; Churko, J.M.; Sallam, K.I.; Matsa, E.; Sharma, A.; et al. Epigenetic Regulation of Phosphodiesterases 2A and 3A Underlies Compromised β-Adrenergic Signaling in an iPSC Model of Dilated Cardiomyopathy. Cell Stem Cell 2015, 17, 89–100. [Google Scholar] [CrossRef]
- Schreier, T.; Kedes, L.; Gahlmann, R. Cloning, structural analysis, and expression of the human slow twitch skeletal muscle/cardiac troponin C gene. J. Biol. Chem. 1990, 265, 21247–21253. [Google Scholar] [CrossRef] [PubMed]
- Gahlmann, R.; Kedes, L. Cloning, structural analysis, and expression of the human fast twitch skeletal muscle troponin C gene. J. Biol. Chem. 1990, 265, 12520–12528. [Google Scholar] [CrossRef]
- Gahlmann, R.; Wade, R.; Gunning, P.; Kedes, L. Differential expression of slow and fast skeletal muscle troponin C. J. Mol. Biol. 1988, 201, 379–391. [Google Scholar] [CrossRef]
- Collins, J.H.; Greaser, M.L.; Potter, J.D.; Horn, M.J. Determination of the amino acid sequence of troponon C from rabbit skeletal muscle. J. Biol. Chem. 1977, 252, 6356–6362. [Google Scholar] [CrossRef]
- Kobayashi, T.; Takagi, T.; Konishi, K.; Morimoto, S.; Ohtsuki, I. Amino Acid Sequence of Porcine Cardiac Muscle Troponin CJ. Biochem. 1989, 106, 55–59. [Google Scholar] [CrossRef]
- Parmacek, M.S.; Leiden, J.M. Structure and Expression of the Murine Slow/Cardiac Troponin C Gene. J. Biol. Chem. 1989, 264, 13217–13225. [Google Scholar] [CrossRef]
- Ding, X.-L.; Akella, Á. B.; Su, H.; Gulati, J. , The role of glycine (residue 89) in the central helix of EF-hand protein troponin-C exposed following amino-terminal α-helix deletion. Protein Sci 1994, 3, 2089–96. [Google Scholar]
- Smith, L.; Greenfield, N.J.; Hitchcock-DeGregori, S.E. Mutations in the N- and D-Helices of the N-Domain of Troponin C Affect the C-Domain and Regulatory Function. Biophys. J. 1999, 76, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Chandra, M.; da Silva, E.; Sorenson, M.; Ferro, J.; Pearlstone, J.; Nash, B.; Borgford, T.; Kay, C.; Smillie, L. The effects of N helix deletion and mutant F29W on the Ca2+ binding and functional properties of chicken skeletal muscle troponin. J. Biol. Chem. 1994, 269, 14988–14994. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Greenfield, N.; Hitchcock-DeGregori, S. The effects of deletion of the amino-terminal helix on troponin C function and stability. J. Biol. Chem. 1994, 269, 9857–9863. [Google Scholar] [CrossRef]
- Herzberg, O.; James, M.N.G. Structure of the calcium regulatory muscle protein troponin-C at 2.8 Å resolution. Nature 1985, 313, 653–659. [Google Scholar] [CrossRef]
- Herzberg, O.; James, M.N. G, Refined crystal structure of troponin C from turkey skeletal muscle at 2. 0 Å resolution. J. Mol. Biol. 1988, 203, 761–779. [Google Scholar] [CrossRef] [PubMed]
- A Satyshur, K.; Rao, S.T.; Pyzalska, D.; Drendel, W.; Greaser, M.; Sundaralingam, M. Refined structure of chicken skeletal muscle troponin C in the two-calcium state at 2-A resolution. J. Biol. Chem. 1988, 263, 1628–1647. [Google Scholar] [CrossRef]
- Putkey, J.; Liu, W.; Sweeney, H. Function of the N-terminal calcium-binding sites in cardiac/slow troponin C assessed in fast skeletal muscle fibers. 2021, 266, 14881–14884. [Google Scholar] [CrossRef]
- Slupsky, C. M.; Sykes, B. D. , NMR solution structure of calcium-saturated skeletal muscle troponin C. Biochemistry 1995, 34, 15953–64. [Google Scholar] [CrossRef]
- Houdusse, A.; Love, M. L.; Dominguez, R.; Grabarek, Z.; Cohen, C. , Structures of four Ca2+-bound troponin C at 2. 0 Å resolution: further insights into the Ca2+-switch in the calmodulin superfamily. Structure 1997, 5, 1695–711. [Google Scholar]
- Sia, S.K.; Li, M.X.; Spyracopoulos, L.; Gagné, S.M.; Liu, W.; Putkey, J.A.; Sykes, B.D. Structure of Cardiac Muscle Troponin C Unexpectedly Reveals a Closed Regulatory Domain. J. Biol. Chem. 1997, 272, 18216–18221. [Google Scholar] [CrossRef]
- Takeda, S.; Yamashita, A.; Maeda, K.; Maéda, Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 2003, 424, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Bers, D. M. , Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar]
- Westerblad, H.; Allen, D.G. Relaxation, [Ca2+]i and [Mg2+]i during prolonged tetanic stimulation of intact, single fibres from mouse skeletal muscle. J. Physiol. 1994, 480, 31–43. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Brinkmeier, H.; Müntener, M. Calcium Ion in Skeletal Muscle: Its Crucial Role for Muscle Function, Plasticity, and Disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [CrossRef] [PubMed]
- Kushmerick, M.J.; Dillon, P.F.; A Meyer, R.; Brown, T.R.; Krisanda, J.M.; Sweeney, H.L. 31P NMR spectroscopy, chemical analysis, and free Mg2+ of rabbit bladder and uterine smooth muscle. J. Biol. Chem. 1986, 261, 14420–14429. [Google Scholar] [CrossRef] [PubMed]
- Godt, R.E.; Maughan, D.W. On the composition of the cytosol of relaxed skeletal muscle of the frog. Am. J. Physiol. Physiol. 1988, 254, C591–C604. [Google Scholar] [CrossRef]
- Homsher, E.; Kean, C. J. , Skeletal muscle energetics and metabolism. Annu. Rev. Physiol. 1978, 40, 93–131. [Google Scholar]
- Hou, T.T.; Johnson, J.D.; A Rall, J. Parvalbumin content and Ca2+ and Mg2+ dissociation rates correlated with changes in relaxation rate of frog muscle fibres. J. Physiol. 1991, 441, 285–304. [Google Scholar] [CrossRef]
- Potter, J.D.; Gergely, J. The calcium and magnesium binding sites on troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 1975, 250, 4628–4633. [Google Scholar] [CrossRef]
- Negele, J.C.; Dotson, D.G.; Liu, W.; Sweeney, H.L.; A Putkey, J. Mutation of the high affinity calcium binding sites in cardiac troponin C. J. Biol. Chem. 1992, 267, 825–831. [Google Scholar] [CrossRef]
- Badr, M. A.; Pinto, J. R.; Davidson, M. W.; Chase, P. B. , Fluorescent protein-based Ca2+ sensor reveals global, divalent cation-dependent conformational changes in cardiac troponin C. PLoS One 2016, 11, e0164222. [Google Scholar]
- Davis, J.P.; Rall, J.A.; Reiser, P.J.; Smillie, L.B.; Tikunova, S.B.; Wesche, J.; Małecki, J.; Więdłocha, A.; Skjerpen, C.S.; Claus, P.; et al. Engineering Competitive Magnesium Binding into the First EF-hand of Skeletal Troponin C. J. Biol. Chem. 2002, 277, 49716–49726. [Google Scholar] [CrossRef]
- Skowronsky, R.A.; Schroeter, M.; Baxley, T.; Li, Y.; Chalovich, J.M.; Spuches, A.M. Thermodynamics and molecular dynamics simulations of calcium binding to the regulatory site of human cardiac troponin C: evidence for communication with the structural calcium binding sites. JBIC J. Biol. Inorg. Chem. 2012, 18, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Rayani, K.; Seffernick, J.; Li, A.Y.; Davis, J.P.; Spuches, A.M.; Van Petegem, F.; Solaro, R.J.; Lindert, S.; Tibbits, G.F. Binding of calcium and magnesium to human cardiac troponin C. J. Biol. Chem. 2021, 296, 100350. [Google Scholar] [CrossRef]
- van Eerd, J.-P.; Takahashi, K. , Determination of the complete amino acid sequence of bovine cardiac troponin C. Biochemistry 1976, 15, 1171–1180. [Google Scholar]
- Sweeney, H.L.; Brito, R.M.; Rosevear, P.R.; A Putkey, J. The low-affinity Ca2(+)-binding sites in cardiac/slow skeletal muscle troponin C perform distinct functions: site I alone cannot trigger contraction. Proc. Natl. Acad. Sci. 1990, 87, 9538–9542. [Google Scholar] [CrossRef] [PubMed]
- Marques, M. d. A.; Pinto, J. R.; Moraes, A. H.; Iqbal, A.; de Magalhães, M. T. Q.; Monteiro, J.; Pedrote, M. M.; Sorenson, M. M.; Silva, J. L.; de Oliveira, G. A. P. , Allosteric Transmission along a Loosely Structured Backbone Allows a Cardiac Troponin C Mutant to Function with Only One Ca2+ Ion. J. Biol. Chem. 2017, 292, 2379–2394. [Google Scholar]
- Vinogradova, M. V.; Stone, D. B.; Malanina, G. G.; Karatzaferi, C.; Cooke, R.; Mendelson, R. A.; Fletterick, R. J. , Ca2+-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA 2005, 102, 5038–43. [Google Scholar]
- Ikura, M.; Clore, G. M.; Gronenborn, A. M.; Zhu, G.; Klee, C. B.; Bax, A. , Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 1992, 256, 632–638. [Google Scholar]
- Collins, J.H. Myosin light chains and troponin C: Structural and evolutionary relationships revealed by amino acid sequence comparisons. J. Muscle Res. Cell Motil. 1991, 12, 3–25. [Google Scholar] [CrossRef]
- Houdusse, A.; Silver, M.; Cohen, C. A model of Ca2+-free calmodulin binding to unconventional myosins reveals how calmodulin acts as a regulatory switch. Structure 1996, 4, 1475–1490. [Google Scholar] [CrossRef]
- Gillis, T. E.; Marshall, C. R.; Tibbits, G. F. , Functional and evolutionary relationships of troponin C. Physiol. Genomics 2007, 32, 16–27. [Google Scholar] [PubMed]
- Shi, Y.; Bethea, J. P.; Hetzel-Ebben, H. L.; Landim-Vieira, M.; Mayper, R. J.; Williams, R. L.; Kessler, L. E.; Ruiz, A. M.; Gargiulo, K.; Rose, J. S. M.; Platt, G.; Pinto, J. R.; Washburn, B. K.; Chase, P. B. , Mandibular muscle troponin of the Florida carpenter ant Camponotus floridanus: extending our insights into invertebrate Ca2+ regulation. J. Muscle Res. Cell Motil. 2021, 42, 399–417. [Google Scholar] [CrossRef]
- Agianian, B.; Kržič, U.; Qiu, F.; A Linke, W.; Leonard, K.; Bullard, B. A troponin switch that regulates muscle contraction by stretch instead of calcium. EMBO J. 2004, 23, 772–779. [Google Scholar] [CrossRef]
- Qiu, F.; Lakey, A.; Agianian, B.; Hutchings, A.; Butcher, G.W.; Labeit, S.; Leonard, K.; Bullard, B. Troponin C in different insect muscle types: identification of two isoforms in Lethocerus, Drosophila and Anopheles that are specific to asynchronous flight muscle in the adult insect. Biochem. J. 2003, 371, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.H.; Theibert, J.L.; Francois, J.M.; Ashley, C.C.; Potter, J.D. Amino acid sequences and calcium-binding properties of two isoforms of barnacle troponin C. Biochemistry 1991, 30, 702–707. [Google Scholar] [CrossRef] [PubMed]
- van Eerd, J.-P.; Takahashi, K. , The amino acid sequence of bovine cardiac troponin-C. Comparison with rabbit skeletal troponin-C. Biochem. Biophys. Res. Commun. 1975, 64, 122–127. [Google Scholar] [CrossRef]
- Wilkinson, J.M. Troponin C from Rabbit Slow Skeletal and Cardiac Muscle Is the Product of a Single Gene. Eur. J. Biochem. 1980, 103, 179–188. [Google Scholar] [CrossRef]
- Roher, A.; Lieska, N.; Spitz, W. The amino acid sequence of human cardiac troponin-C. Muscle Nerve 1986, 9, 73–77. [Google Scholar] [CrossRef]
- Wilkinson, J. The amino acid sequence of troponin C from chicken skeletal muscle. FEBS Lett. 1976, 70, 254–256. [Google Scholar] [CrossRef]
- Romero-Herrera, A. E.; Castillo, O.; Lehmann, H. , Human skeletal muscle proteins. The primary structure of troponin C. J. Mol. Evol. 1976, 8, 251–70. [Google Scholar] [PubMed]
- Strasburg, G.; Greaser, M.; Sundaralingam, M. X-ray diffraction studies of troponin-C crystals from rabbit and chicken skeletal muscles. J. Biol. Chem. 1980, 255, 3806–3808. [Google Scholar] [CrossRef]
- McKay, R.T.; Tripet, B.P.; Hodges, R.S.; Sykes, B.D. Interaction of the Second Binding Region of Troponin I with the Regulatory Domain of Skeletal Muscle Troponin C as Determined by NMR Spectroscopy. 1997; 272, 2849. [Google Scholar] [CrossRef]
- Mckay, R.; Pearlstone, J.; Corson, D.; Gagne, S.; Smillie, L.; Sykes, B. ; Structure and interaction site of the regulatory domain of troponin-C when complexed with the 96-148 region of troponin-I. Biochemistry 1998, 37, 12419–30. [Google Scholar]
- McKay, R.T.; Tripet, B.P.; Pearlstone, J.R.; Smillie, L.B.; Sykes, B.D. Defining the Region of Troponin-I that Binds to Troponin-C. Biochemistry 1999, 38, 5478–5489. [Google Scholar] [CrossRef] [PubMed]
- Li, M. X.; Spyracopoulos, L.; Sykes, B. D. , Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry 1999, 38, 8289–98. [Google Scholar]
- Vassylyev, D.G.; Takeda, S.; Wakatsuki, S.; Maeda, K.; Maéda, Y. Crystal structure of troponin C in complex with troponin I fragment at 2.3-Å resolution. Proc. Natl. Acad. Sci. 1998, 95, 4847–4852. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Takeda, S.; Scherer, A.; Kobayashi, T.; Maeda, Y.; Taniguchi, H.; Yao, M.; Wakatsuki, S. , Production, crystallization, and preliminary X-ray analysis of rabbit skeletal muscle troponin complex consisting of troponin C and fragment (1-47) of troponin I. Protein Sci 1997, 6, 916–8. [Google Scholar]
- Findlay, W.A.; Sykes, B.D. Proton NMR resonance assignments, secondary structure, and global fold of the TR1C fragment of turkey skeletal troponin C in the calcium-free state. Biochemistry 1993, 32, 3461–3467. [Google Scholar] [CrossRef]
- Li, M.X.; Gagne, S.M.; Tsuda, S.; Kay, C.M.; Smillie, L.B.; Sykes, B.D. Calcium Binding to the Regulatory N-Domain of Skeletal Muscle Troponin C Occurs in a Stepwise Manner. Biochemistry 1995, 34, 8330–8340. [Google Scholar] [CrossRef]
- Spyracopoulos, L.; Li, M.X.; Sia, S.K.; Gagné, S.M.; Chandra, M.; Solaro, R.J.; Sykes, B.D. Calcium-Induced Structural Transition in the Regulatory Domain of Human Cardiac Troponin C, Biochemistry 1997, 36, 12138–12146. [Google Scholar] [CrossRef]
- Findlay, W.; Soennichsen, F.; Sykes, B.D. Solution structure of the TR1C fragment of skeletal muscle troponin-C. J. Biol. Chem. 1994, 269, 6773–8. [Google Scholar] [PubMed]
- Oda, T.; Yanagisawa, H.; Wakabayashi, T. Cryo-EM structures of cardiac thin filaments reveal the 3D architecture of troponin. J. Struct. Biol. 2020, 209, 107450. [Google Scholar] [CrossRef]
- Risi, C. M.; Belknap, B.; White, H. D.; Dryden, K.; Pinto, J. R.; Chase, P. B.; Galkin, V. E. , High-resolution cryo-EM structure of the junction region of the native cardiac thin filament in relaxed state. PNAS Nexus 2023, 2, pgac298. [Google Scholar]
- Regnier, M.; Rivera, A.; Chase, P.; Smillie, L.; Sorenson, M. Regulation of Skeletal Muscle Tension Redevelopment by Troponin C Constructs with Different Ca2+ Affinities. Biophys. J. 1999, 76, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Gulati, J.; Babu, A.; Su, H.; Zhang, Y. Identification of the regions conferring calmodulin-like properties to troponin C. J. Biol. Chem. 1993, 268, 11685–11690. [Google Scholar] [CrossRef] [PubMed]
- Pearlstone, J.R.; Borgford, T.; Chandra, M.; Oikawa, K.; Kay, C.M.; Herzberg, O.; Moult, J.; Herklotz, A.; Reinach, F.C.; Smillie, L.B. Construction and characterization of a spectral probe mutant of troponin C: application to analyses of mutants with increased calcium affinity. Biochemistry 1992, 31, 6545–6553. [Google Scholar] [CrossRef]
- da Silva, E.F.; Sorenson, M.M.; Smillie, L.B.; Barrabin, H.; Scofano, H.M. Comparison of calmodulin and troponin C with and without its amino-terminal helix (residues 1-11) in the activation of erythrocyte Ca(2+)-ATPase. J. Biol. Chem. 1993, 268, 26220–26225. [Google Scholar] [CrossRef]
- Babu, A.; Orr, G.; Gulati, J. Calmodulin supports the force-generating function in desensitized muscle fibers. J. Biol. Chem. 1988, 263, 15485–15491. [Google Scholar] [CrossRef]
- Hill, A. V. , The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 1910, 40, iv–vii. [Google Scholar]
- Reinoso, T.R.; Landim-Vieira, M.; Shi, Y.; Johnston, J.R.; Chase, P.B.; Parvatiyar, M.S.; Landstrom, A.P.; Pinto, J.R.; Tadros, H.J. A comprehensive guide to genetic variants and post-translational modifications of cardiac troponin C. J. Muscle Res. Cell Motil. 2020, 42, 323–342. [Google Scholar] [CrossRef]
- Willott, R. H.; Gomes, A. V.; Chang, A. N.; Parvatiyar, M. S.; Pinto, J. R.; Potter, J. D. , Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 2010, 48, 882–92. [Google Scholar] [CrossRef] [PubMed]
- Tadros, H.J.; Life, C.S.; Garcia, G.; Pirozzi, E.; Jones, E.G.; Datta, S.; Parvatiyar, M.S.; Chase, P.B.; Allen, H.D.; Kim, J.J.; et al. Meta-analysis of cardiomyopathy-associated variants in troponin genes identifies loci and intragenic hot spots that are associated with worse clinical outcomes. J. Mol. Cell. Cardiol. 2020, 142, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Na, I.; Kong, M. J.; Straight, S.; Pinto, J. R.; Uversky, V. N. , Troponins, intrinsic disorder, and cardiomyopathy. Biol Chem 2016, 397, 731–51. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Parvatiyar, M.S.; Pinto, J.R.; Marquardt, M.L.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Potter, J.D.; Ackerman, M.J. Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 2008, 45, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Parvatiyar, M.S.; Jones, M.A.; Liang, J.; Ackerman, M.J.; Potter, J.D. A Functional and Structural Study of Troponin C Mutations Related to Hypertrophic Cardiomyopathy. J. Biol. Chem. 2009, 284, 19090–19100. [Google Scholar] [CrossRef]
- Swindle, N.; Tikunova, S.B. Hypertrophic Cardiomyopathy-Linked Mutation D145E Drastically Alters Calcium Binding by the C-Domain of Cardiac Troponin C. Biochemistry 2010, 49, 4813–4820. [Google Scholar] [CrossRef]
- Pinto, J. R.; Reynaldo, D. P.; Parvatiyar, M. S.; Dweck, D.; Liang, J.; Jones, M. A.; Sorenson, M. M.; Potter, J. D. , Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. J. Biol. Chem. 2011, 286, 1005–13. [Google Scholar] [CrossRef]
- Albury, A.N.J.; Swindle, N.; Swartz, D.R.; Tikunova, S.B. Effect of Hypertrophic Cardiomyopathy-Linked Troponin C Mutations on the Response of Reconstituted Thin Filaments to Calcium upon Troponin I Phosphorylation. Biochemistry 2012, 51, 3614–3621. [Google Scholar] [CrossRef]
- Martins, A.S.; Parvatiyar, M.S.; Turna, R.; Badger, C.D.; Griffin, B.; Zorio, D.; Vukmirovic, M.; Sanchez-Gonzalez, M.A.; Dweck, D.; Ruiz, E.L.; et al. In Vivo Analysis of Troponin C Knock-In (A8V) Mice: Evidence that TNNC1 is a Hypertrophic Cardiomyopathy Susceptibility Gene. Biophys. J. 2014, 106, 723a–723A. [Google Scholar] [CrossRef]
- Kathuria, S. V.; Chan, Y. H.; Nobrega, R. P.; Özen, A.; Matthews, C. R. , Clusters of isoleucine, leucine, and valine side chains define cores of stability in high-energy states of globular proteins: Sequence determinants of structure and stability. Protein Sci 2016, 25, 662–75. [Google Scholar]
- Zot, H.G.; Hasbun, J.E.; Michell, C.A.; Landim-Vieira, M.; Pinto, J.R. Enhanced troponin I binding explains the functional changes produced by the hypertrophic cardiomyopathy mutation A8V of cardiac troponin C. Arch. Biochem. Biophys. 2016, 601, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Baxley, T.; Johnson, D.; Pinto, J. R.; Chalovich, J. M. , Troponin C Mutations Partially Stabilize the Active State of Regulated Actin and Fully Stabilize the Active State When Paired with Δ14 TnT. Biochemistry 2017, 56, 2928–2937. [Google Scholar] [PubMed]
- Kawai, M.; Johnston, J.R.; Karam, T.; Wang, L.; Singh, R.K.; Pinto, J.R. Myosin Rod Hypophosphorylation and CB Kinetics in Papillary Muscles from a TnC-A8V KI Mouse Model. Biophys. J. 2017, 112, 1726–1736. [Google Scholar] [CrossRef]
- Stevens, C.M.; Rayani, K.; Singh, G.; Lotfalisalmasi, B.; Tieleman, D.; Tibbits, G.F. Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J. Biol. Chem. 2017, 292, 11915–11926. [Google Scholar] [CrossRef] [PubMed]
- Veltri, T.; Landim-Vieira, M.; Parvatiyar, M.S.; Gonzalez-Martinez, D.; Jones, K.M.D.; Michell, C.A.; Dweck, D.; Landstrom, A.P.; Chase, P.B.; Pinto, J.R. Hypertrophic Cardiomyopathy Cardiac Troponin C Mutations Differentially Affect Slow Skeletal and Cardiac Muscle Regulation. Front. Physiol. 2017, 8, 221. [Google Scholar] [CrossRef]
- Gonzalez-Martinez, D.; Johnston, J.R.; Landim-Vieira, M.; Ma, W.; Antipova, O.; Awan, O.; Irving, T.C.; Chase, P.B.; Pinto, J.R. Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell. Cardiol. 2018, 123, 26–37. [Google Scholar] [CrossRef]
- Johnston, J.R.; Landim-Vieira, M.; Marques, M.A.; de Oliveira, G.A.; Gonzalez-Martinez, D.; Moraes, A.H.; He, H.; Iqbal, A.; Wilnai, Y.; Birk, E.; et al. The intrinsically disordered C terminus of troponin T binds to troponin C to modulate myocardial force generation. J. Biol. Chem. 2019, 294, 20054–20069. [Google Scholar] [CrossRef]
- Jones, K.M.D.; Vied, C.; Valera, I.C.; Chase, P.B.; Parvatiyar, M.S.; Pinto, J.R. Sexual dimorphism in cardiac transcriptome associated with a troponin C murine model of hypertrophic cardiomyopathy. Physiol. Rep. 2020, 8, e14396. [Google Scholar] [CrossRef]
- E de Feria, A.; E Kott, A.; Becker, J.R. Sarcomere mutation negative hypertrophic cardiomyopathy is associated with ageing and obesity. Open Hear. 2021, 8, e001560. [Google Scholar] [CrossRef]
- Pinto, J.R.; Siegfried, J.D.; Parvatiyar, M.S.; Li, D.; Norton, N.; Jones, M.A.; Liang, J.; Potter, J.D.; Hershberger, R.E. Functional Characterization of TNNC1 Rare Variants Identified in Dilated Cardiomyopathy. J. Biol. Chem. 2011, 286, 34404–34412. [Google Scholar] [CrossRef]
- Hespe, S.; Waddell, A.; Asatryan, B.; Owens, E.; Thaxton, C.; Adduru, M.-L.; Anderson, K.; Brown, E. E.; Hoffman-Andrews, L.; Jordan, E.; Josephs, K.; Mayers, M.; Peters, S.; Stafford, F.; Bagnall, R. D.; Bronicki, L.; Callewaert, B.; Chahal, C. A. A.; James, C. A.; Jarinova, O.; Landstrom, A. P.; McNally, E. M.; Murray, B.; Muiño-Mosquera, L.; Parikh, V.; Reuter, C.; Walsh, R.; Wayburn, B.; Ware, J. S.; Ingles, J. , Genes Associated with Hypertrophic Cardiomyopathy: A Reappraisal by the ClinGen Hereditary Cardiovascular Disease Gene Curation Expert Panel. J. Am. Coll. Cardiol. 2025, 85, 727–740. [Google Scholar] [PubMed]
- Juárez, C.K.; Sequeira, V.; Boogaard, M.v.D.; Veerman, C.C.; Hoetjes, N.J.; Poel, E.; Tanck, M.W.; Deprez, R.H.L.; Vermeer, A.M.; van der Velden, J.; et al. Tropomyosin–troponin complex in inherited cardiomyopathies. Hear. Rhythm. 2024, 21, 1173–1175. [Google Scholar] [CrossRef]
- Tikunova, S.B.; Thuma, J.; Davis, J.P. Mouse Models of Cardiomyopathies Caused by Mutations in Troponin C. Int. J. Mol. Sci. 2023, 24, 12349. [Google Scholar] [CrossRef]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Enciso, J.S.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef]
- Hassoun, R.; Budde, H.; Mannherz, H.G.; Lódi, M.; Fujita-Becker, S.; Laser, K.T.; Gärtner, A.; Klingel, K.; Möhner, D.; Stehle, R.; et al. De Novo Missense Mutations in TNNC1 and TNNI3 Causing Severe Infantile Cardiomyopathy Affect Myofilament Structure and Function and Are Modulated by Troponin Targeting Agents. Int. J. Mol. Sci. 2021, 22, 9625. [Google Scholar] [CrossRef]
- Tobacman, L.S.; Cammarato, A. Cardiomyopathic troponin mutations predominantly occur at its interface with actin and tropomyosin. J. Gen. Physiol. 2021, 153. [Google Scholar] [CrossRef]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med Genet. Part A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Patsalis, C.; Kyriakou, S.; Georgiadou, M.; Ioannou, L.; Constantinou, L.; Soteriou, V.; Jossif, A.; Evangelidou, P.; Sismani, C.; Kypri, E.; et al. Investigating TNNC1 gene inheritance and clinical outcomes through a comprehensive familial study. Am. J. Med Genet. Part A 2024, 197, e63838. [Google Scholar] [CrossRef]
- Veltri, T.; de Oliveira, G.A.P.; Bienkiewicz, E.A.; Palhano, F.L.; Marques, M.d.A.; Moraes, A.H.; Silva, J.L.; Sorenson, M.M.; Pinto, J.R. Amide hydrogens reveal a temperature-dependent structural transition that enhances site-II Ca2+-binding affinity in a C-domain mutant of cardiac troponin C. Sci. Rep. 2017, 7, 691. [Google Scholar] [CrossRef]
- Marques, M.A.; Landim-Vieira, M.; Moraes, A.H.; Sun, B.; Johnston, J.R.; Jones, K.M.D.; Cino, E.A.; Parvatiyar, M.S.; Valera, I.C.; Silva, J.L.; et al. Anomalous structural dynamics of minimally frustrated residues in cardiac troponin C triggers hypertrophic cardiomyopathy. Chem. Sci. 2021, 12, 7308–7323. [Google Scholar] [CrossRef]
- Varughese, J.F.; Baxley, T.; Chalovich, J.M.; Li, Y. A Computational and Experimental Approach To Investigate Bepridil Binding with Cardiac Troponin. J. Phys. Chem. B 2011, 115, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- van de Locht, M.; Donkervoort, S.; de Winter, J.M.; Conijn, S.; Begthel, L.; Kusters, B.; Mohassel, P.; Hu, Y.; Medne, L.; Quinn, C.; et al. Pathogenic variants in TNNC2 cause congenital myopathy due to an impaired force response to calcium. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Garcia, M.R.; Schmeckpeper, J.; Landim-Vieira, M.; Coscarella, I.L.; Fang, X.; Ma, W.; Spran, P.A.; Yuan, S.; Qi, L.; Kahmini, A.R.; et al. Disruption of Z-Disc Function Promotes Mechanical Dysfunction in Human Myocardium: Evidence for a Dual Myofilament Modulatory Role by Alpha-Actinin Int. J. Mol. Sci. 2023, 24, 14572. [Google Scholar] [CrossRef]
- Landim-Vieira, M.; Childers, M.C.; Wacker, A.L.; Garcia, M.R.; He, H.; Singh, R.; A Brundage, E.; Johnston, J.R.; A Whitson, B.; Chase, P.B.; et al. Post-translational modification patterns on β-myosin heavy chain are altered in ischemic and nonischemic human hearts. eLife 2022, 11. [Google Scholar] [CrossRef]
- Sevrieva, I.; Knowles, A.C.; Kampourakis, T.; Sun, Y.-B. Regulatory domain of troponin moves dynamically during activation of cardiac muscle. J. Mol. Cell. Cardiol. 2014, 75, 181–187. [Google Scholar] [CrossRef]
- Sevrieva, I.R.; Kampourakis, T.; Irving, M. Structural changes in troponin during activation of skeletal and heart muscle determined in situ by polarised fluorescence. Biophys. Rev. 2024, 16, 753–772. [Google Scholar] [CrossRef]
Figure 3.
Partial polypeptide sequence alignment of sTnC (upper) and cTnC (lower) of Homo sapiens. The first 12 amino acids are highlighted in yellow. Note that the N-terminal Met is typically removed post-translationally from sTnC but not cTnC, followed by acetylation of the remaining N-terminal amino group. Identical amino acids are indicated below the two sequences by an asterisk (*), conserved substitutions by a colon (:), and semi-conserved substitutions by a period (.). The alignment was obtained using UniProt Clustal Omega.
Figure 3.
Partial polypeptide sequence alignment of sTnC (upper) and cTnC (lower) of Homo sapiens. The first 12 amino acids are highlighted in yellow. Note that the N-terminal Met is typically removed post-translationally from sTnC but not cTnC, followed by acetylation of the remaining N-terminal amino group. Identical amino acids are indicated below the two sequences by an asterisk (*), conserved substitutions by a colon (:), and semi-conserved substitutions by a period (.). The alignment was obtained using UniProt Clustal Omega.

Figure 4.
The N-helix of cTnC (magenta) is centrally located among the cTnC D-helix, D/E-linker and cTnI helix H1 in all Ca2+-free (CF; pCa 8) and Ca2+-bound (CB; pCa 4) structural models of portions of the cardiac thin filament [22]. One major distinction between CF and CB states is the location of the carboxyl terminus of cTnI (blue structure at top of each panel): in CF states, it extends away (upward in the panels) from the core domain of troponin along tropomyosin (green) and actin (gray), while in CB states, part of the cTnI sequence is bound to the N-lobe of cTnC. Another major distinction between CF and CB states is the location of tropomyosin (green) relative to actin (gray); displacement of tropomyosin (green) exposes myosin-binding sites on actin (gray) in CB states, permitting actomyosin crossbridge cycling (not shown) and contraction. Panels A, B and C are CF (PDB: 8UWW), CF-tilted (CF-T) (PDB: 8UWX) and CF-rotated (CF-R) (PDB: 8UWY) models of the upper strand, respectively, while panels D, E and F refer to the corresponding models of the lower strand. The -tilted and -rotated designations refer to the orientation of the IT-arm (blue-yellow α-helical coiled-coil that is central to each panel) relative to that in the CF state. The upper strand has two CB states: CB-partially-activated (CB-PA) (G; PDB: 8UZY) and CB-fully-activated (CB-FA) (H; PDB: 8UZX). The lower strand also has CB-PA state (I; PDB: 8V0K), but two different CB-FA states (J & K; PDB: 8V01 & 8V0I) are captured. In this figure, actin is gray; tropomyosin is green; cTnI is blue; cTnT is yellow; and the cTnC subunit is red with the N-helix sequence highlighted in magenta. Note the marked change in orientation of the N-helix between Ca2+-free (A-F) and Ca2+-bound (G-K) states.
Figure 4.
The N-helix of cTnC (magenta) is centrally located among the cTnC D-helix, D/E-linker and cTnI helix H1 in all Ca2+-free (CF; pCa 8) and Ca2+-bound (CB; pCa 4) structural models of portions of the cardiac thin filament [22]. One major distinction between CF and CB states is the location of the carboxyl terminus of cTnI (blue structure at top of each panel): in CF states, it extends away (upward in the panels) from the core domain of troponin along tropomyosin (green) and actin (gray), while in CB states, part of the cTnI sequence is bound to the N-lobe of cTnC. Another major distinction between CF and CB states is the location of tropomyosin (green) relative to actin (gray); displacement of tropomyosin (green) exposes myosin-binding sites on actin (gray) in CB states, permitting actomyosin crossbridge cycling (not shown) and contraction. Panels A, B and C are CF (PDB: 8UWW), CF-tilted (CF-T) (PDB: 8UWX) and CF-rotated (CF-R) (PDB: 8UWY) models of the upper strand, respectively, while panels D, E and F refer to the corresponding models of the lower strand. The -tilted and -rotated designations refer to the orientation of the IT-arm (blue-yellow α-helical coiled-coil that is central to each panel) relative to that in the CF state. The upper strand has two CB states: CB-partially-activated (CB-PA) (G; PDB: 8UZY) and CB-fully-activated (CB-FA) (H; PDB: 8UZX). The lower strand also has CB-PA state (I; PDB: 8V0K), but two different CB-FA states (J & K; PDB: 8V01 & 8V0I) are captured. In this figure, actin is gray; tropomyosin is green; cTnI is blue; cTnT is yellow; and the cTnC subunit is red with the N-helix sequence highlighted in magenta. Note the marked change in orientation of the N-helix between Ca2+-free (A-F) and Ca2+-bound (G-K) states.
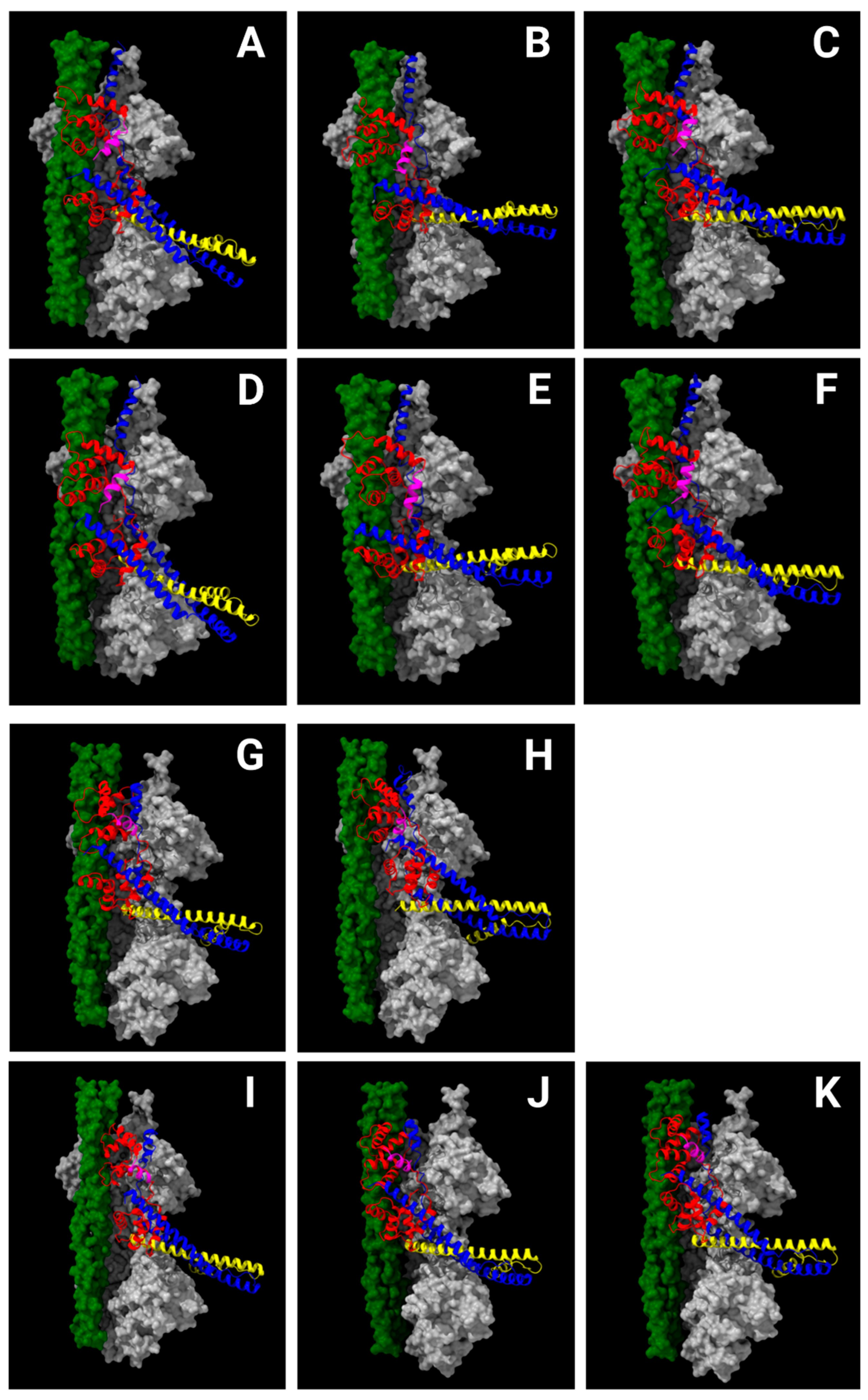
Figure 5.
Location of cardiomyopathy-related residues in N-helix of cTnC. Top: Tertiary structure model of cTnC of upper strand in the CB-FA state (Source: PDB 8UZX) [22]. Bottom: The primary and secondary structure representation of cTnC N-helix (amino acids 2-12). The six affected residues have seven possible cardiomyopathy-related variants, with the ones published in peer-reviewed journals marked with asterisks [112]. The residues relevant to different types of cardiomyopathies are indicated by distinct colors: lime (DCM/HCM), cyan (DCM), yellow (HCM/RCM) and blue (DCM/HCM/RCM).
Figure 5.
Location of cardiomyopathy-related residues in N-helix of cTnC. Top: Tertiary structure model of cTnC of upper strand in the CB-FA state (Source: PDB 8UZX) [22]. Bottom: The primary and secondary structure representation of cTnC N-helix (amino acids 2-12). The six affected residues have seven possible cardiomyopathy-related variants, with the ones published in peer-reviewed journals marked with asterisks [112]. The residues relevant to different types of cardiomyopathies are indicated by distinct colors: lime (DCM/HCM), cyan (DCM), yellow (HCM/RCM) and blue (DCM/HCM/RCM).
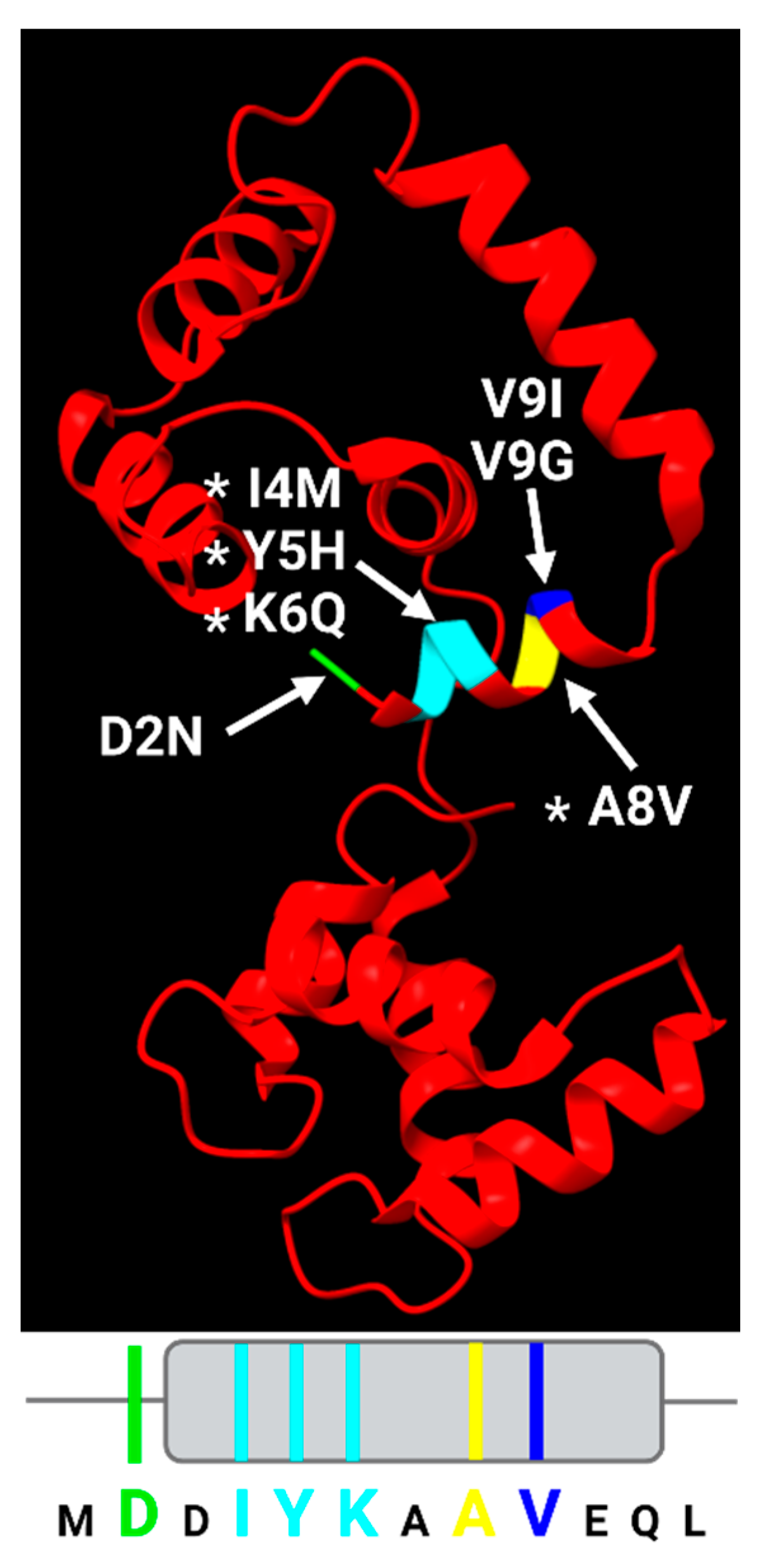
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



