Submitted:
06 March 2025
Posted:
07 March 2025
You are already at the latest version
Abstract
Homology, or relationship among characters by common descent, has been notoriously difficult to assess for many morphological features, and cell types in particular. The ontogenetic origin of such traits means that the only physically inherited information is encoded in the genomes. However, the complexity of the underlying gene regulatory network and often miniscule changes that can impact gene expression, make it practically impossible to postulate a clear demarcation line for what molecular signature should “define” a homologous cell type between two deeply branching animals. In this Hypothesis article, we propose the use of the recently characterized irreversible genomic states, that occur after chromosomal and sub-chromosomal mixing of genes and regulatory elements, to dissect regulatory signatures of each cell type into irreversible and reversible configurations. While many of such states will be non-functional, some may permanently impact gene expression in a given cell type. Our proposal is that such evolutionarily irreversible, and thus synapomorphic, functional genomic states can constitute a criterion for the timing of the origin of deep evolutionary cell type homologies. Our proposal thus aims to close the gap between the clearly defined homology of the individual genomic characters and their genomic states to the homology at the phenotypic level through the identification of the underlying evolutionarily irreversible and regulatory linked states.
Keywords:
chromosome evolution
; enhancer-promoter contacts
; fusion-with-mixing
; entropy
; irreversibility
Homology at the organismal level, including developmental processes [1,2], organ and cell type homology [3,4,5] has been one of the most discussed and debated concepts in the field of evolution and development [6]. At the molecular level, lately fueled by the technological advancement in single cell transcriptomics, the “molecular signature” of many morphological or developmental traits could be identified [4,7,8,9,10,11]. However, complex interplay between regulatory networks during development [12] and the transcription factor logic associated with cell types [4] makes it difficult to identify a clear shared set of genes that identify a given cell type for clades that have been separated from each other for hundreds of millions of years.
High genomic evolvability and complexity of genomic features that can impact gene regulatory networks and their phenotypic outcomes is, arguably, the main unresolved problem when attempting to identify the mechanistic basis of homologies at the organismal level and among distantly related clades. Many genomic changes, from small nucleotide substitutions, enhancer evolution to duplications of genes and regulatory sites [13,14,15] can lead to evolutionary novelty. However, these changes can also equally lead to loss of such novel characters. It is often unclear how often such events can or have occurred on the vast macro-evolutionary (above clade-level) time-scale, and how often regulatory wiring can be reversed to the ancestral states [16]. Furthermore, it has been found that the genetic basis for clearly homologous characters can be different, in particular in terms of its inducing factors during development [17], while the core identity mechanisms are much more conserved [18].
At the 64th Phyletic Symposium in Jena last year, the lively discussion highlighted again the particular problems when assessing the homology of phenotypes and developmental processes that comprise the hourglass model [10,19,20,21]. Ideally, a clear signature of a cell type or a developmental process rooted in the underlying genomic information needs to be identified.
In this Hypothesis we argue for a possible link between genome structure and the cell type homology at the macro-evolutionary level. We propose the identification of irreversible or highly “entangled” genomic states [22] that may affect gene expression and that can be related to the origin of a particular cell type or cell type family. We suggest that irreversibility is the key property in any identification of distant organismal homologies, as, considering the macro-evolutionary time-scales, there is otherwise no theoretical boundary to (re)evolve various gene expression patterns. Only gene regulation that is linked to a specific irreversible genomic state can be treated as a stable synapomorphic character within a given clade. Evolutionary irreversibility of a given genomic configuration also means that without the information from the outgroup species about the possible ancestral state, we irrevocably lose this ancestral state information.
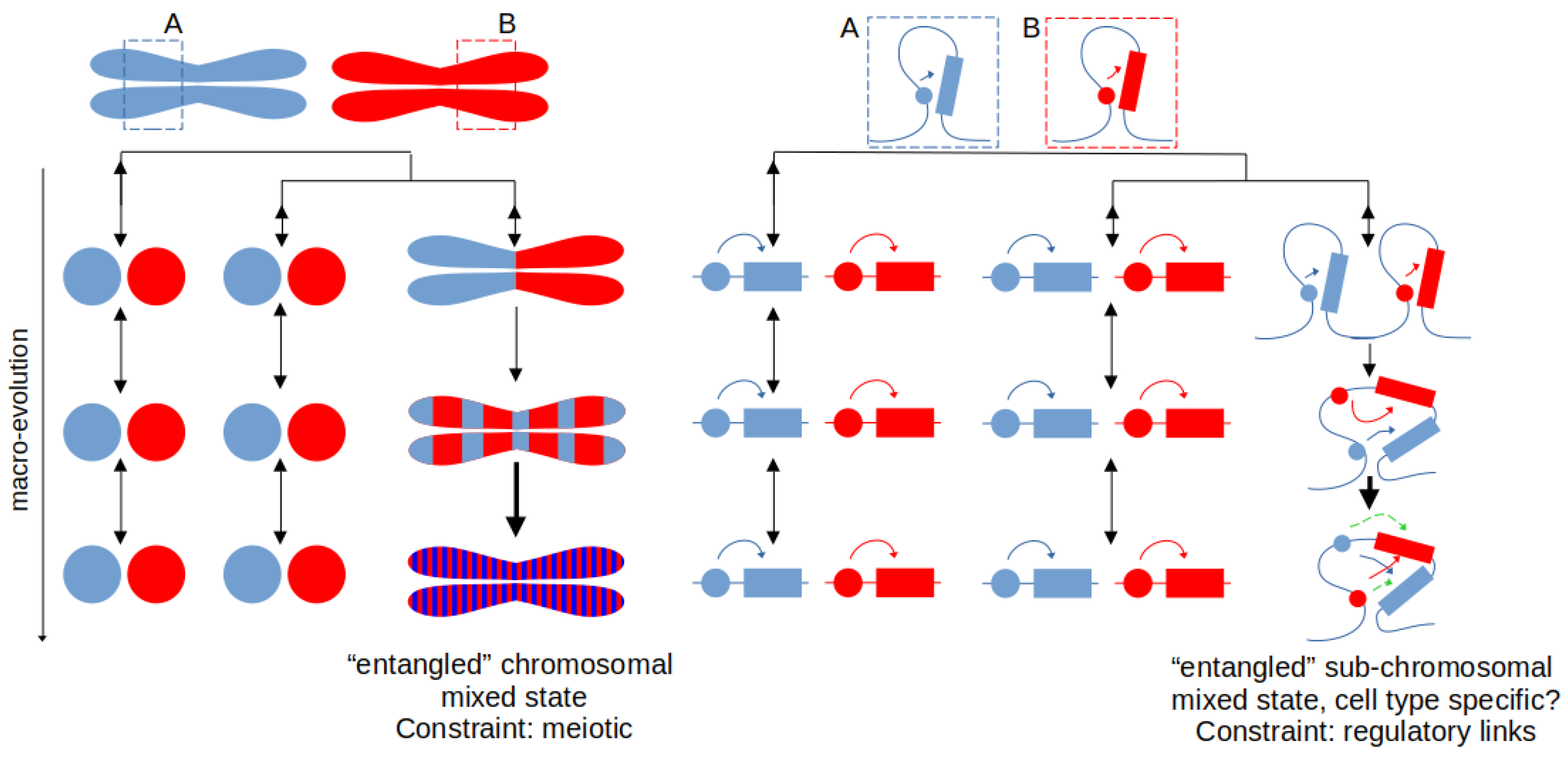
Over the recent years, accumulation of chromosomal-scale genomes has enabled us to trace the evolution of whole chromosomes and study their composition as a function of orthologous gene families [23,24]. One of the key identified properties for animal chromosomal evolution was the so-called “fusion-with-mixing” (Figure 1): when two ancestrally conserved chromosomes undergo fusion, the genes on them mix through intra-chromosomal translocations and the resulting state (mixed chromosome) cannot be reverted to the original two states comprised of the two ancestral gene complements [23]. This chromosomal-scale mixing thus constitutes a very strong synapomorphic character that, once established, cannot be reverted and is expected to be observed in all descendants of a particular lineage. This property has already been utilized to shed new light onto highly debated phylogenetic positions [25].
The observed maintenance of chromosomal-scale linkages can be largely explained by meiotic constraints [26,27,28] and so far little evidence exists of any regulatory function [29]. On the other hand, how genes explore their local, sub-chromosomal, interactive environments and the impact of this process for the evolution of novel gene regulation has already been suggested (e.g., the addition of hundreds of novel topological and co-regulated units after large-scale genome rearrangements [30]). More recent insights suggest that, due to the ongoing process of intra-chromosomal translocations, hundreds of stable regulatory interactions can emerge even within fully retained (unfused) chromosomes since their origin in the metazoan ancestor [22]. The constraint for the maintenance of such sub-chromosomal linkages can be diverse. Mixing of enhancer-promoter (E-P) interactions within a topologically-interacting space (e.g., via loop-extrusion mechanisms and/or topologically associating domains, TADs [31,32]) may create an “entangled” configuration that is very unlikely to be unmixed by random inversions, as these would otherwise break functional E-P contacts (Figure 1). This constraint thus leads to mixing that is analogous to the chromosomal “fusion-with-mixing”, but on a much smaller, sub-chromosomal, level. Thus, similar to the deeply conserved chromosomal-level synteny, such constraints may result in retention of unrelated genes and their regulatory regions within specific genomic neighborhoods (Figure 1). This prediction is corroborated both by the frequently observed micro-synteny in animal genomes [33,34,35,36,37,38] as well as by the findings that translocations usually happen at the TAD boundaries [39]. The resulting maintenance of local linkages does not imply an immediate functional advantage, with any potential synergetic function evolving after this initial “entanglement” (Figure 1). It can thus be expected that this process can lead to the evolution and maintenance of a more complex enhancer logic within or associated with the local syntenic neighborhood.
The suggested irreversibility of this evolutionary process enables us to link such states to specific changes in gene expression and, eventually, to gene expression that is associated with cell type development or function. It suggests that gene sets that define each cell type or a developmental stage can be analyzed in terms of their synapomorphic states or regulatory entanglements. Phylogenetic dating of the regulatory entanglements and quantification of their irreversibility in particular can indicate at what evolutionary node the novelty arose and that a scenario of re-ancestralization (unmixing, “disentanglement”) can be ruled out. It will also be possible to define the probability of convergence of such mixed states, as has been done for chromosomal-level fusion-with-mixing events, which will depend on the number of the involved genomic elements that undergo mixing [24,25].
The exact quantification and the methodology of the identification of such states begins to emerge [22], building on novel interdisciplinary applications, including topological theories [40] in macro-evolution. Such ideas may establish a fruitful testing ground for many deep evolutionary phenotype homology hypotheses. A signature of such entangled states would comprise of a set of enhancers and their target gene(s) located within one interactive region (e.g., a TAD or a loop) where the homologous regions in multiple outgroup species are located either in separate interactive environments or on different chromosomes [30] (Figure 1). The changes in the gene expression associated with a particular cell type should also be tested in this context and it should be expected that regulatory entanglement would create a more complex regulatory logic, e.g., by creating additive sub-functionalized enhancers or entangled super-enhancers within that region [41,42]. Furthermore, regulatory entanglement may also lead to the accumulation of several such states within the same genomic locus, i.e., two already entangled states undergoing further fusion and mixing. An example may constitute the vertebrate HoxD cluster and complex regulation of the C-TAD and T-TAD regions around it, compared to the plesiomorphic invertebrate state [38,43,44,45]. While complex enhancer logic in many systems and loci has been reported, including enhancers that act over larger genomic distances [46], the determination of how much of it arose through regulatory entanglement is lacking. It is important to note that, similar to chromosomal rearrangement events in some clades that break the ancestral metazoan chromosomal homologies [47], sub-chromosomally “mixed” clusters such as Hox can also break apart and scramble within or between chromosomes (e.g., Hox cluster scrambling in some metazoan lineages [34,48,49]). However, as at the chromosomal level, sub-chromosomal “fission” products are not homologous to the ancestral pre-fusion or pre-mixing state, i.e., they can not theoretically revert back to any of the proposed ancestral Hox configurations [50].
In general, this logic may be viewed to some degree similar to what has been described for phylostratigraphy approaches using orphan (novel) genes and other genomic changes that are evolutionary very rare [51,52]. However, novel genes often do not perform key cell type associated function, and, by definition, have no homology relationship to the outgroups where those genes are not found. Thus, such characters have limited implications for ancient cell type origination. In this context, it is also important to note that we do not propose that irreversible states resulting from mixing of genes and regulatory elements are the only driving force in phenotype or specifically cell type evolution. Clearly, many studies have shown a tremendous multitude of genomic changes that can result in changes in the gene expression. For core regulatory complexes (CoRCs), co-evolution and co-adaptation among transcription factor proteins has been proposed which may comprise a novel “mixed” state [4,53]. However, without a clear understanding of transition and reversibility properties of such changes across macro-evolution their implications for the cell type constituting molecular signatures are limited. We can thus envisage the next macro-evolutionary genomics frontier that will encompass studying irreversibility properties in the evolution of a plethora of such states. One such additional and least explored feature is the continuing expansion and integration of transposable elements into existing regulatory landscapes, which, through enlargement of the E-P distances as well as origin of novel regulatory sites, may facilitate formation of cell-type-associated irreversible states [54,55] and lead to their “fossilization” [56,57].
In summary, we propose that to be able to identify homology at the macro-evolutionary scale, irreversibility of the underlying genomic states, if present, may comprise a key criterion. We argue that positional characters and their regulatory “entanglement” along chromosomes may provide a testable concept to help address this long-lasting question. Finally, knowledge and phylogenetic dating of entangled genomic states will be useful in directing sequencing efforts of species that may still retain some of the ancestral un-mixed configurations, helping quantify how much of the ancestral information has been lost due to this irreversible mixing.
Funding
O.S. was supported by the European Research Council’s Horizon 2020: European Union Research and Innovation Programme, grant No. 945026. GPW acknowledges research funding from the Hagler Institute for Advanced Studies at Texas A&M University.
Acknowledgments
Authors would like to thank all members of the Simakov lab, Ludwik Gąsiorowski, and Eve Seuntjens for valuable discussions and input on the manuscript.
References
- Baer KE von. Über Entwicklungsgeschichte der Thiere. 1. Auflage. Königsberg: Bornträger; 1828.
- Hall, B.K. Phylotypic stage or phantom: is there a highly conserved embryonic stage in vertebrates? Trends Ecol. Evol. 1997, 12, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Valentine JW. Cell Types, Numbers, and Body Plan Complexity. In: Hall BK, editor. Keywords and Concepts in Evolutionary Developmental Biology [Internet]. Harvard University Press; 2006 [cited 2024 Dec 2]. p. 35–43. Available from: https://www.degruyter.com/document/doi/10.4159/9780674273320-008/html.
- Arendt, D.; Musser, J.M.; Baker, C.V.H.; Bergman, A.; Cepko, C.; Erwin, D.H.; Pavlicev, M.; Schlosser, G.; Widder, S.; Laubichler, M.D.; et al. The origin and evolution of cell types. Nat. Rev. Genet. 2016, 17, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Wagner GP. Homology, Genes, and Evolutionary Innovation. 1st edition. Princeton: Princeton University Press; 2014.
- Gould SJ. Ontogeny and phylogeny. Harvard University Press; 1985.
- Sebé-Pedrós, A.; Chomsky, E.; Pang, K.; Lara-Astiaso, D.; Gaiti, F.; Mukamel, Z.; Amit, I.; Hejnol, A.; Degnan, B.M.; Tanay, A. Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat. Ecol. Evol. 2018, 2, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Sebé-Pedrós, A.; Saudemont, B.; Chomsky, E.; Plessier, F.; Mailhé, M.-P.; Renno, J.; Loe-Mie, Y.; Lifshitz, A.; Mukamel, Z.; Schmutz, S.; et al. Cnidarian Cell Type Diversity and Regulation Revealed by Whole-Organism Single-Cell RNA-Seq. Cell 2018, 173, 1520–1534.e20. [Google Scholar] [CrossRef]
- Martín-Durán, J.M.; Hejnol, A. A developmental perspective on the evolution of the nervous system. Dev. Biol. 2021, 475, 181–192. [Google Scholar] [CrossRef]
- Kalinka, A.T.; Varga, K.M.; Gerrard, D.T.; Preibisch, S.; Corcoran, D.L.; Jarrells, J.; Ohler, U.; Bergman, C.M.; Tomancak, P. Gene expression divergence recapitulates the developmental hourglass model. Nature 2010, 468, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Paganos P, Voronov D, Musser J, Arendt D, Arnone MI. Single cell RNA sequencing of the Strongylocentrotus purpuratus larva reveals the blueprint of major cell types and nervous system of a non-chordate deuterostome [Internet]. bioRxiv; 2021 [cited 2022 Dec 5]. p. 2021.03.16.435574. Available from: https://www.biorxiv.org/content/10.1101/2021.03.16.435574v3.
- Davidson, E.H.; Erwin, D.H. Gene regulatory networks and the evolution of animal body plans. Science 2006, 311, 796–797. [Google Scholar] [CrossRef]
- Touceda-Suárez, M.; Kita, E.M.; Acemel, R.D.; Firbas, P.N.; Magri, M.S.; Naranjo, S.; Tena, J.J.; Gómez-Skarmeta, J.L.; Maeso, I.; Irimia, M. Ancient Genomic Regulatory Blocks Are a Source for Regulatory Gene Deserts in Vertebrates after Whole-Genome Duplications. Mol. Biol. Evol. 2020, 37, 2857–2864. [Google Scholar] [CrossRef]
- Sommer-Trembo, C.; Santos, M.E.; Clark, B.; Werner, M.; Fages, A.; Matschiner, M.; Hornung, S.; Ronco, F.; Oliver, C.; Garcia, C.; et al. The genetics of niche-specific behavioral tendencies in an adaptive radiation of cichlid fishes. Science 2024, 384, 470–475. [Google Scholar] [CrossRef]
- Villar, D.; Berthelot, C.; Aldridge, S.; Rayner, T.F.; Lukk, M.; Pignatelli, M.; Park, T.J.; Deaville, R.; Erichsen, J.T.; Jasinska, A.J.; et al. Enhancer Evolution across 20 Mammalian Species. Cell 2015, 160, 554–566. [Google Scholar] [CrossRef]
- Babonis, L.S.; Enjolras, C.; Reft, A.J.; Foster, B.M.; Hugosson, F.; Ryan, J.F.; Daly, M.; Martindale, M.Q. Single-cell atavism reveals an ancient mechanism of cell type diversification in a sea anemone. Nat. Commun. 2023, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McColgan, Á.; DiFrisco, J. Understanding developmental system drift. Development 2024, 151. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P. The developmental genetics of homology. Nat. Rev. Genet. 2007, 8, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Haeckel E, Haeckel E. Generelle morphologie der organismen. Allgemeine grundzüge der organischen formen-wissenschaft, mechanisch begründet durch die von Charles Darwin reformirte descendenztheorie [Internet]. Berlin: G. Reimer; 1866. Available from: https://www.biodiversitylibrary.org/bibliography/3953.
- Levit, G.S.; Hoßfeld, U.; Naumann, B.; Lukas, P.; Olsson, L. The biogenetic law and the Gastraea theory: From Ernst Haeckel's discoveries to contemporary views. J. Exp. Zoöl. Part B: Mol. Dev. Evol. 2021, 338, 13–27. [Google Scholar] [CrossRef]
- Uesaka, M.; Kuratani, S.; Irie, N. The developmental hourglass model and recapitulation: An attempt to integrate the two models. J. Exp. Zoöl. Part B: Mol. Dev. Evol. 2021, 338, 76–86. [Google Scholar] [CrossRef]
- Schultz DT, Blümel A, Destanović D, Sarigol F, Simakov O. Topological mixing and irreversibility in animal chromosome evolution. bioRxiv. 2024;2024–07.
- Simakov, O.; Marlétaz, F.; Yue, J.-X.; O’connell, B.; Jenkins, J.; Brandt, A.; Calef, R.; Tung, C.-H.; Huang, T.-K.; Schmutz, J.; et al. Deeply conserved synteny resolves early events in vertebrate evolution. Nat. Ecol. Evol. 2020, 4, 820–830. [Google Scholar] [CrossRef]
- Simakov, O.; Bredeson, J.; Berkoff, K.; Marletaz, F.; Mitros, T.; Schultz, D.T.; O’connell, B.L.; Dear, P.; Martinez, D.E.; Steele, R.E.; et al. Deeply conserved synteny and the evolution of metazoan chromosomes. Sci. Adv. 2022, 8, eabi5884. [Google Scholar] [CrossRef]
- Schultz, D.T.; Haddock, S.H.D.; Bredeson, J.V.; Green, R.E.; Simakov, O.; Rokhsar, D.S. Ancient gene linkages support ctenophores as sister to other animals. Nature 2023, 618, 110–117. [Google Scholar] [CrossRef]
- Muller HJ. Bearing of the “Drosophila” work on systematics. The New Systematics [Internet]. 1940 [cited 2021 Feb 8]. p. 185–268. Available from: https://ci.nii.ac.jp/naid/10004957361/.
- Wright, S. On the Probability of Fixation of Reciprocal Translocations. Am. Nat. 1941, 75, 513–522. [Google Scholar] [CrossRef]
- Lv, J.; Havlak, P.; Putnam, N.H. Constraints on genes shape long-term conservation of macro-synteny in metazoan genomes. BMC Bioinform. 2011, 12, S11–S11. [Google Scholar] [CrossRef]
- Clarence, T.; Robert, N.S.; Sarigol, F.; Fu, X.; Bates, P.A.; Simakov, O. Robust 3D modeling reveals spatiosyntenic properties of animal genomes. iScience 2023, 26, 106136. [Google Scholar] [CrossRef] [PubMed]
- Schmidbaur, H.; Kawaguchi, A.; Clarence, T.; Fu, X.; Hoang, O.P.; Zimmermann, B.; Ritschard, E.A.; Weissenbacher, A.; Foster, J.S.; Nyholm, S.V.; et al. Emergence of novel cephalopod gene regulation and expression through large-scale genome reorganization. Nat. Commun. 2022, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szalay, M.-F.; Majchrzycka, B.; Jerković, I.; Cavalli, G.; Ibrahim, D.M. Evolution and function of chromatin domains across the tree of life. Nat. Struct. Mol. Biol. 2024, 31, 1824–1837. [Google Scholar] [CrossRef]
- Rogers, T.F.; Simakov, O. Emerging questions on the mechanisms and dynamics of 3D genome evolution in spiralians. Briefings Funct. Genom. 2023, 22, 533–542. [Google Scholar] [CrossRef]
- Irimia, M.; Tena, J.J.; Alexis, M.S.; Fernandez-Miñan, A.; Maeso, I.; Bogdanović, O.; de la Calle-Mustienes, E.; Roy, S.W.; Gómez-Skarmeta, J.L.; Fraser, H.B. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Res. 2012, 22, 2356–2367. [Google Scholar] [CrossRef]
- Simakov, O.; Marletaz, F.; Cho, S.-J.; Edsinger-Gonzales, E.; Havlak, P.; Hellsten, U.; Kuo, D.-H.; Larsson, T.; Lv, J.; Arendt, D.; et al. Insights into bilaterian evolution from three spiralian genomes. Nature 2012, 493, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Engström, P.G.; Sui, S.J.H.; Drivenes, Ø.; Becker, T.S.; Lenhard, B. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Res. 2007, 17, 1898–1908. [Google Scholar] [CrossRef]
- Kikuta, H.; Laplante, M.; Navratilova, P.; Komisarczuk, A.Z.; Engström, P.G.; Fredman, D.; Akalin, A.; Caccamo, M.; Sealy, I.; Howe, K.; et al. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Res. 2007, 17, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, B.; Robert, N.S.M.; Technau, U.; Simakov, O. Ancient animal genome architecture reflects cell type identities. Nat. Ecol. Evol. 2019, 3, 1289–1293. [Google Scholar] [CrossRef]
- Robert, N.S.M.; Sarigol, F.; Zimmermann, B.; Meyer, A.; Voolstra, C.R.; Simakov, O. Emergence of distinct syntenic density regimes is associated with early metazoan genomic transitions. BMC Genom. 2022, 23, 1–14. [Google Scholar] [CrossRef]
- Álvarez-González, L.; Burden, F.; Doddamani, D.; Malinverni, R.; Leach, E.; Marín-García, C.; Marín-Gual, L.; Gubern, A.; Vara, C.; Paytuví-Gallart, A.; et al. 3D chromatin remodelling in the germ line modulates genome evolutionary plasticity. Nat. Commun. 2022, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Erwin, D.H. The topology of evolutionary novelty and innovation in macroevolution. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160422. [Google Scholar] [CrossRef]
- Choi, J.; Lysakovskaia, K.; Stik, G.; Demel, C.; Söding, J.; Tian, T.V.; Graf, T.; Cramer, P. Evidence for additive and synergistic action of mammalian enhancers during cell fate determination. eLife 2021, 10. [Google Scholar] [CrossRef]
- Rickels, R.; Shilatifard, A. Enhancer Logic and Mechanics in Development and Disease. Trends Cell Biol. 2018, 28, 608–630. [Google Scholar] [CrossRef] [PubMed]
- Montavon, T.; Soshnikova, N.; Mascrez, B.; Joye, E.; Thevenet, L.; Splinter, E.; de Laat, W.; Spitz, F.; Duboule, D. A Regulatory Archipelago Controls Hox Genes Transcription in Digits. Cell 2011, 147, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carballo, E.; Lopez-Delisle, L.; Willemin, A.; Beccari, L.; Gitto, S.; Mascrez, B.; Duboule, D. Chromatin topology and the timing of enhancer function at the HoxD locus. Proc. Natl. Acad. Sci. 2020, 117, 31231–31241. [Google Scholar] [CrossRef]
- Acemel, R.D.; Tena, J.J.; Irastorza-Azcarate, I.; Marlétaz, F.; Gómez-Marín, C.; de la Calle-Mustienes, E.; Bertrand, S.; Diaz, S.G.; Aldea, D.; Aury, J.-M.; et al. A single three-dimensional chromatin compartment in amphioxus indicates a stepwise evolution of vertebrate Hox bimodal regulation. Nat. Genet. 2016, 48, 336–341. [Google Scholar] [CrossRef]
- Batut, P.J.; Bing, X.Y.; Sisco, Z.; Raimundo, J.; Levo, M.; Levine, M.S. Genome organization controls transcriptional dynamics during development. Science 2022, 375, 566–570. [Google Scholar] [CrossRef]
- Albertin, C.B.; Medina-Ruiz, S.; Mitros, T.; Schmidbaur, H.; Sanchez, G.; Wang, Z.Y.; Grimwood, J.; Rosenthal, J.J.C.; Ragsdale, C.W.; Simakov, O.; et al. Genome and transcriptome mechanisms driving cephalopod evolution. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Ikuta, T.; Yoshida, N.; Satoh, N.; Saiga, H. Ciona intestinalis Hox gene cluster: Its dispersed structure and residual colinear expression in development. Proc. Natl. Acad. Sci. 2004, 101, 15118–15123. [Google Scholar] [CrossRef]
- Seo, H.-C.; Edvardsen, R.B.; Maeland, A.D.; Bjordal, M.; Jensen, M.F.; Hansen, A.; Flaat, M.; Weissenbach, J.; Lehrach, H.; Wincker, P.; et al. Hox cluster disintegration with persistent anteroposterior order of expression in Oikopleura dioica. Nature 2004, 431, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Butts, T.; Holland, P.W.; Ferrier, D.E. The Urbilaterian Super-Hox cluster. Trends Genet. 2008, 24, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Brajković, J.; Tautz, D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007, 23, 533–539. [Google Scholar] [CrossRef]
- Bleidorn, C. Rare Genomic Changes. In Phylogenomics: An Introduction [Internet]; Bleidorn, C., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 195–211. [Google Scholar] [CrossRef]
- Park, Y.; Nnamani, M.C.; Maziarz, J.; Wagner, G.P. Cis-Regulatory Evolution of Forkhead Box O1 (FOXO1), a Terminal Selector Gene for Decidual Stromal Cell Identity. Mol. Biol. Evol. 2016, 33, 3161–3169. [Google Scholar] [CrossRef] [PubMed]
- Lynch, V.J.; Leclerc, R.D.; May, G.; Wagner, G.P. Transposon-mediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet. 2011, 43, 1154–1159. [Google Scholar] [CrossRef]
- Kon-Nanjo K, Kon T, Yu TC-TK, Rodriguez-Terrones D, Falcon F, Martínez DE, et al. The dynamic genomes of Hydra and the anciently active repeat complement of animal chromosomes [Internet]. bioRxiv; 2024 [cited 2024 Nov 12]. p. 2024.03.13.584568. Available from: https://www.biorxiv.org/content/10.1101/2024.03.13.584568v1.
- Meyer, A.; Schloissnig, S.; Franchini, P.; Du, K.; Woltering, J.M.; Irisarri, I.; Wong, W.Y.; Nowoshilow, S.; Kneitz, S.; Kawaguchi, A.; et al. Giant lungfish genome elucidates the conquest of land by vertebrates. Nature 2021, 590, 284–289. [Google Scholar] [CrossRef]
- Schartl, M.; Woltering, J.M.; Irisarri, I.; Du, K.; Kneitz, S.; Pippel, M.; Brown, T.; Franchini, P.; Li, J.; Li, M.; et al. The genomes of all lungfish inform on genome expansion and tetrapod evolution. Nature 2024, 634, 96–103. [Google Scholar] [CrossRef]
Figure 1.
Irreversible genomic states at chromosomal (left) and sub-chromosomal (right) scales, their origin, and occasional function. Left: chromosomal fusion-with-mixing occurs via, e.g., Robertsonian translocation, followed by intra-chromosomal inversions. The information about the ancestral two states (two separate chromosomes) is lost after the mixing and if no plesiomorphic (outgroup) information is available. Unfused chromosomal states in the “outgroup” species are shown as circles, due to space constraints. Right: similar mixing can be observed for the more functionally relevant enhancer-promoter (E-P) contacts within a single chromosome (labelled as region “A” and region “B”, with E-P link shown in blue and red, respectively). E-P links are mixed within an interactive environment (mediated, e.g., through DNA loop-extrusion) making it unlikely for random inversions to disentangle them into the original state without breaking functional E-P contacts. Over longer time-scales, this “entanglement” may lead to the evolution of novel (green arrows) persistent E-P links. Black vertical arrows indicate possible evolutionary transitions between homologous states (two-way arrow: reversible; single-way arrow: irreversible). Slightly thicker single-way arrow for the final mixed state suggests higher level of its entropic mixing.
Figure 1.
Irreversible genomic states at chromosomal (left) and sub-chromosomal (right) scales, their origin, and occasional function. Left: chromosomal fusion-with-mixing occurs via, e.g., Robertsonian translocation, followed by intra-chromosomal inversions. The information about the ancestral two states (two separate chromosomes) is lost after the mixing and if no plesiomorphic (outgroup) information is available. Unfused chromosomal states in the “outgroup” species are shown as circles, due to space constraints. Right: similar mixing can be observed for the more functionally relevant enhancer-promoter (E-P) contacts within a single chromosome (labelled as region “A” and region “B”, with E-P link shown in blue and red, respectively). E-P links are mixed within an interactive environment (mediated, e.g., through DNA loop-extrusion) making it unlikely for random inversions to disentangle them into the original state without breaking functional E-P contacts. Over longer time-scales, this “entanglement” may lead to the evolution of novel (green arrows) persistent E-P links. Black vertical arrows indicate possible evolutionary transitions between homologous states (two-way arrow: reversible; single-way arrow: irreversible). Slightly thicker single-way arrow for the final mixed state suggests higher level of its entropic mixing.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



