
Submitted:
07 March 2025
Posted:
07 March 2025
You are already at the latest version
Abstract
In humans, aging is an inevitable consequence of diminished growth processes after being full-grown. The high order of biological material in cells and tissues is continuously disturbed by numerous physical and chemical destructive impacts. Host-derived oxidant-based cytotoxic agents (reactive species, transition free metal ions, free heme) contribute considerably to this damage. These agents are under control by immediately acting antagonizing principles, which are important to ensure cell and tissue homeostasis. During aging, energy metabolism and supply of tissues with dioxygen and nutrients are increasingly disturbed. In addition, a chronic inflammatory state is developing, a condition known as inflammaging. In this review, the interplay between oxidant-based cytotoxic agents and protective mechanisms is analyzed in dependence on age-based physiological alterations in ATP production. Disturbances in this balance are associated with the development of age-related diseases and comorbidities. The enhanced production of reactive species from dysfunctional mitochondria, alterations in cellular redox homeostasis, and adaptations to hypoxia are highlighted. Examples are given how disturbances between oxidant-based cytotoxic agents and antagonizing principles contribute to the pathogenesis of diseases in persons of advanced age.
Keywords:
aging
; cytotoxic agents
; antagonizing principles
; inflammaging
; hypoxia
; redox homeostasis
; oxidative stress
; reactive species
; transition metal ions
; free heme
1. Introduction
Aging is accompanied by a gradual decline of physiological functions and physical activity and is associated with increased predisposition to different health-threatening diseases. Despite the existence of about 300 theories and hypothesis about the reasons of aging, there does not exist any comprehensive description explaining all miraculous facets of the aging process [1]. These theories on aging can be roughly divided into three main categories: i) theories related to deviations in description of genetic information, ii) theories favoring any problems in hormone metabolism, and iii) theories based on accumulation of waste products in cells and the organism [2]. Although numerous facts are provided in favors of all these theories, it remains unknown how these different approaches can be synergized in a unifying theory.
In attempt to create a more complex trial of description of the aging process, I proposed that aging is an inevitable consequence of growth limitation during the development of complex multicellular organisms [2,3]. On the one hand, in a growing organism, cells are dividing permanently, increasing thus the mass of the biological material. In order to supply adequately all cells of this organism with energy and food, differentiation and functional specification of cells and formation of specific tissue types and organs are mandatory. This further increases the complexity of the developing organism. On the other hand, each kind of biological material is devoid to numerous chemical and physical impacts that can disturb the integrity of this highly ordered living matter and impairs thus physiological processes in the growing organism. Despite the existence of numerous immediately acting and inducible protective mechanisms in living matter, only a sufficient high growth rate can ensure functionally intact organisms. After reaching an optimum size for each organism, the general growth rate declines more and more. As a result, destructive elements can’t be compensated as efficiently as before [3]. There is also a gradual shift in energy balance in elderly organisms from organism-specific processes like active movement, sensing the environment, food intake, or physical and mental work towards the predominance of processes counterbalancing any deviations from normal physiological functioning on systemic and also cellular level [2]. With increasing age, more and more energy equivalents are required to maintain essential physiological parameters in cells and tissues of the organism.
In organisms, important protective functions are realized by the immune and associated systems like acute-phase, complement, coagulation, and contact systems [4,5]. The activity of these systems is directed to ensure the maintenance of basic homeostatic parameters in cells, tissues, and the whole organism. The steady increase of any kind of destructions in elderly persons enhances the activity of these protective systems and is associated with an increase of permanent inflammatory states and immune dysfunctions. This condition, which is known as inflammaging [6], represents a key risk factor for the increased appearance of life-threatening chronic diseases and adverse health outcomes [7].
During inflammatory response several host-derived cytotoxic agents are generated and released from activated immune cells and undergoing tissue cells [8]. These agents contribute to destruction and elimination of foreign microorganisms and unwanted cells, but can also disturb the integrity of healthy cells and tissues. Cytotoxic agents are usually well controlled by corresponding antagonizing principles to avoid any damage of unperturbed cells in close neighborhood to inflammatory sites. Disturbances in the balance between cytotoxic agents and protecting mechanisms favor chronic inflammatory states and disease progression [8]. Otherwise, overexpression of antagonizing principles as found for example in late-stage tumor cells promotes also markedly the disease process [9].
In this review, the interplay between oxidant-based host-derived cytotoxic agents and protective mechanisms is analyzed in elderly persons. The question will be addressed how the increased appearance of inflammatory states in aged individuals affects this balance and contributes to pathogenesis of life-threatening diseases.
2. Key Concepts About Inflammation in Aged Individuals
2.1. The Concept of Inflammaging
The concept of inflammaging was developed from the network hypothesis of aging [10,11], which is based on the idea that in elderly individuals the ability of different anti-stress systems is limited to counterbalance any destructive deviations in cells and organs of elderly organisms. A key point of inflammaging is that aging is associated with an increased activity of both the innate and activated immune responses in elderly persons [12]. Indeed, with advanced age, the serum level of a variety of pro-inflammatory mediators increases as shown for IL-6, IL-15, IL-8 [12,13,14], and coagulation factors like fibrinogen and von Willebrand factor [15,16]. In total, this increase indicates the presence of a permanent inflammatory state that can be identified as a low-grade chronic inflammation [17].
According to the inflammaging hypothesis, the balance between pro-inflammatory and anti-anti-inflammatory agents is disturbed in aged individuals [12]. This balance is crucial for resistance against any kind of diseases. Important anti-inflammatory agents in elderly persons are TGF-β, cortisol, IL-10, and lipoxins [12,18,19].
Without going into details, the concept of inflammaging summarizes key features characteristic of the chronic inflammatory process in aged persons [7,12,17,20,21,22,23,24]. To get an answer why an inflammation persists under these conditions, it is necessary to analyze how molecular patterns and host-derived cytotoxic agents contribute to the development of long-lasting inflammatory states.
2.2. Molecular Patterns and Host-Derived Cytotoxic Agents in Inflammation
During inflammation, the activation of the immune system and associated protective mechanisms is mandatory to eliminate pathogens, transformed and damaged cells and to induce repair processes. In humans, inflammatory response is initiated by the presence of molecular patterns, which activate immune cascades via interaction with pattern recognition receptors (PRRs) [25]. These molecules result from pathogens (pathogen-associated molecular patterns (PAMPs)) and undergoing immune and non-immune cells of the host (damage-associated molecular pattern, DAMPs) [26,27]. Important DAMPs are heat-shock proteins, high-mobility group box 1 protein, fragments of hyaluronan, uric acid, heparan sulfate, free heme, and tumor DNA [28,29,30,31,32,33]. An acute inflammation is terminated when the number of molecular patterns becomes beneath an unrecognizable level. In other words, inflammation becomes resolved when the pathogen load is considerably reduced and the damage of host cells is substantially limited.
Concerning major mediators and pathways, an acute inflammation can be subdivided into two main phases, initiation and propagation of inflammation as well as resolution of inflammation. During the first phase of an acute inflammation, as a result of the presence of molecular patterns several immune cells are recruited to inflammatory sites and activated, whereby pro-inflammatory cytokines like IL-1, IL-6, IL-8, IL-15, tumor-necrosis factor α (TNF-α) are involved in attraction of neutrophils, monocytes, macrophages, T-cells, and others [34,35,36]. In addition, several acute-phase proteins are released into circulation, at which C-reactive protein (CRP) and serum amyloid A (SAA) serve as important biomarkers of the course of inflammation [34,37,38]. At inflamed loci, immune cells combat against pathogens, and remove damaged cells and cell debris. In macrophages (here subtype M1) and other immune cells, glycolysis is the preferred pathway for ATP production [39,40,41,42]. In the second phase of an acute inflammation, immune cells are suppressed. There is now a dominance of repair processes and synthesis of novel extracellular matrix components. These processes are driven by resolving mediators like transforming growth factor β (TGF-β), IL-10, vascular endothelial growth factor (VEGF), and lipoxins and directed to restore the former tissue homeostasis [43,44,45,46,47,48,49]. The number of myeloid-derived suppressor cells (MDSCs) increases markedly [50,51]. The macrophage subtype M2, which actively promotes oxidative phosphorylation, dominates now [52,53]. Key events, mediators, and pathways of both main phases of an acute inflammation are summarized in Table 1.
By which mechanisms, resolution of inflammation is initiated at inflammatory sites remains puzzling. Under discussion are the enhanced formation of specific resolving mediators like TGF-β, IL-10, VEGF, and lipoxins, which drive accumulated immune cells into an anti-inflammatory and immunosuppressive state and promote tissue repair processes [43,44,45,46,47,48,49]. Another aspect concerns the prevailing dominance of apoptotic cell death processes over the uncontrolled release of cytotoxic agents from necrotic tissues and activated immune cells at inflammatory loci. In addition, release of lactate from activated pro-inflammatory macrophages (M1 type) can induce in neighbored yet unperturbed invading macrophages a metabolic switch towards the M2 type [54,55,56,57]. Lactate serves as fuel in these cells to drive mitochondrial energy metabolism.
In order to better understand how an acute inflammatory response becomes persistent, it is necessary to analyze the role of different host-derived cytotoxic agents in these processes [8]. Molecular patterns are able to initiate inflammatory events. Recruited and activated immune cells are directed to inactivate and kill pathogens and transformed cells, and to destroy and remove damaged cell material. In these activities, immune cells apply different cytotoxic agents, which can also damage yet unperturbed cells at inflammatory sites. Healthy cells and tissues are equipped with numerous antagonizing principles, which inactivate immediately cytotoxic agents and limit thus their unwanted destructive actions. Cytotoxic products of activated neutrophils and their antagonizing principles are listed in Table 2. Other major sources for cytotoxic agents are other immune cells, defective mitochondria, blood hemorrhages, and damaged muscles. In addition to cytotoxic agents listed in Table 2, other important cytotoxic agents are peroxynitrite, free heme, lipid hydroperoxides, mast cell proteases, angiotensin, bradykinin, and matrix metalloproteases [8].
The interplay between cytotoxic agents and antagonizing principles highly determines the further fate of an inflammation [8]. The inflammation becomes resolved when antagonizing principles properly control the action of cytotoxic agents and deactivate them. Any disturbance in the balance between cytotoxic agents and antagonizing principles can result in chronic inflammatory states due to the inability of protective mechanisms to eliminate or inactivate efficiently host-derived cytotoxic agents [8]. As a result of this inability, novel DAMPs and antigens are released by the action of cytotoxic agents and the inflammatory process continues. A scheme about the interplay between molecular patterns and cytotoxic agents in persistence of chronic inflammatory states is given in Figure 1.
In aged people, low-grade chronic inflammation dominates. It is evident that the terms pro-inflammatory and anti-inflammatory agents used in the concept of inflammaging [12] correspond to the predominating mediators in the two main phases of an acute inflammation. During initiation and propagation of inflammation pro-inflammatory agents play an important role and anti-inflammatory mediators are typical of the resolution of inflammation. In analyzing inflammatory processes in aged individuals, the main focus has been directed on the behavior of different pro- and anti-inflammatory markers in association with selected disease processes. Considering the imbalance between cytotoxic agents and antagonizing principles in the development of different age-related disease scenarios, in this review the attention is focused on the role of reactive species, the loss of control over the sequestration of transition metal ions, and diminution and exhaustion of haptoglobin and hemopexin. Oxidant-related cytotoxic agents have a high potential to disturb metabolic processes within cells during aging and to contribute thus to development of disease scenarios associated in aged people.
In analyzing molecular mechanisms of age-related metabolic alterations and conditions for the development of diseases, changes in energy metabolism provide the fundamental clue to better understand the disturbed interplay between oxidant-based cytotoxic agents and antagonizing principles in aged individuals.
3. Peculiarities of the Energy Metabolism and Oxidative Stress in Aged Individuals
3.1. Energy Metabolism Under Normoxic Conditions
In humans and higher animals, adenosine triphosphate (ATP) is the master molecule to provide energy equivalents for numerous energy-intensive processes such as muscle contraction, kinase activities, active transport, intra- and extracellular signaling and others [87,88,89]. In human cells, ATP can be generated both in the cytosol by anerobic glycolysis and most of all within mitochondria by oxidative phosphorylation. Consumption of 1 mole glucose during dioxygen-independent cytosolic glycolysis yields 2 moles ATP. In mitochondria, production of ATP is linked to the tricarboxylic acid (TCA) cycle (Krebs cycle) and the electron transport chain through mitochondrial complexes I-IV. During these processes a pH gradient and an electrical potential are generated across the inner mitochondrial membrane, which are essential for proper functionality of ATP synthase. These metabolic pathways highly depend on the presence of dioxygen and yield about 30-36 moles ATP per mole initial substrate [90].
Several pathways can supply substrates that enter the TCA cycle for subsequent ATP production. Degradation of glucose by glycolysis yields pyruvate and further acetyl-CoA, which enters the TCA cycle. The latter substance is also produced during β-oxidation of long-chain fatty acids. Which type of fuel molecules is utilized for ATP production in cells depends on cell function and physiological context [91]. In cardiac muscle cells, and adipose tissue, β-oxidation of fatty acids predominates to produce ATP. Otherwise, glucose is the preferred fuel molecule in cells of the central nervous system. Degradation of free amino acids contribute to about 10-15 % ATP generated via oxidative phosphorylation [92]. After deamination, the remaining carbon skeleton of amino acids is converted into intermediates, which can enter the TCA cycle at several places.
Although mitochondrial oxidative phosphorylation is more efficient than cytosolic glycolysis concerning the amount of ATP generated per mole initial substrate, glycolysis predominates as main process of energy production in some immune cells (e. g. in activated neutrophils [42], activated macrophages (M1 type) [40], maturating dendritic cells [41], Th1, Th2, Th17 lymphocytes [39,93,94], muscle fibers type IIB [95], and under certain (patho)physiological conditions (e. g. starvation, prolonged exercise). ATP formation by glycolysis is more rapid than within mitochondria. Moreover, glycolysis can be further intensified by up-regulation of the involved enzymes and fast supply of additional glucose from glycogen stores [96].
Starvation, prolonged intense exercise, some diets, dioxygen deprivation, or disease conditions can deplete glycogen stores in liver and muscles [96,97]. As a result, ketone bodies are formed and the de novo synthesis of glucose is accelerated in the liver. Both mechanisms contribute to ATP production. Ketone bodies (acetoacetic acid, acetone, and β-hydroxybutyrate) are derived from fatty acids in the liver and converted after their transport to tissues into acetyl-CoA, which enters the TCA cycle [98]. In the liver and to some extent also in the kidneys, glucose can be generated by gluconeogenesis, whereby lactate, glycerol, glutamine, and alanine are the starting sources for this pathway [92,99]. By these mechanisms, ATP maintenance is ensured to some extent even under short-term and mild disturbances of energy supply and different (patho)physiological conditions are bypassed. Major pathways of cellular energy metabolism are schematically depicted in Figure 2.
3.2. Deviations in Energy Metabolism in Aged Individuals
In aged individuals, production of energy equivalents by oxidative phosphorylation can be highly disturbed by dioxygen deficiency, the presence of dysfunctional mitochondria, and a lower number of these organelles [100,101,102]. Under these conditions, the TCA cycle and mitochondrial electron transport chain are unable to generate high amounts of ATP. During aging, it may occur that glycolysis is highly up-regulated with lactate as final product. Deficient mitochondrial processes can lead to enhanced formation of superoxide anion radicals (O2∙-) and other reactive species like hydrogen peroxide (H2O2), Fe2+, hydroxyl radicals (HO∙), and peroxynitrite (ONOO-), which all are derived from reactions of superoxide anion radicals. Important alterations in energy metabolism in aged individuals are also highlighted in Figure 2.
Several aspects are under discussion contributing to reduced capacity of mitochondria for ATP production in aged individuals. i) Mitochondrial DNA is more prone to damage than nuclear DNA [103]. The lack of histones within mitochondria and a less efficient nucleic acid repair machinery are responsible for the higher mutation rate of mitochondrial DNA [104]. ii) Enhanced production of mitochondria-derived reactive species is another key mediator disturbing enzymes and proteins involved in oxidative phosphorylation. Hypoxic conditions are a major contributor to the increased generation of mitochondria-derived reactive species. iii) In skeletal muscle, the number of mitochondria declines gradually with increasing age [105]. iv) The rate of apoptosis of cells with defective mitochondria increases too with increasing age [106].
Age-related alterations in mitochondria are also reflected by the decline of NAD+ [107,108] and α-ketoglutarate [109,110]. NAD+ is involved in many enzymatic reactions within mitochondria. This decline impairs, for example, the activity of sirtuins, which play an important role in the maintenance of mitochondrial homeostasis [111,112,113,114]. Decreased level of α-ketoglutarate affects the epigenetic landscape in several tissues [110,115,116].
Regarding molecular mechanisms of energy metabolism in mitochondria, accelerated aging is mainly favored by activation of the mTOR pathway and suppression of autophagy by increased formation of aspartate from oxalacetate or increased yield of pyruvate, which is further converted to acetate [117]. These mechanisms are supported by high caloric food and sedentary lifestyle. Further factors contributing to accelerated aging are formation of advanced glycation end products due to enhanced glycolysis rate, impaired repair of DNA hypermethylation induced by increased succinate levels, and reduced maintenance of the stem cell pool by increased level of α-ketoglutarate [117]. Several alterations in the activity of TCA cycle enzymes are responsible for the aforementioned processes. Oxidative stress is associated with decreased activity of succinate dehydrogenase, α-ketoglutarate dehydrogenase, and aconitase and favors the conversion of oxalacetate and glutamate by aspartate aminotransferase into α-ketoglutarate and aspartate. Hence, enhanced levels of aspartate, succinate, and α-ketoglutarate result [117]. Aconitase contains a 4Fe-4S-cluster at active site. Under stress conditions, superoxide anion radicals are known to release Fe2+ from this iron-sulfur cluster [118,119].
Decreased energy expenditure by oxidative phosphorylation can be compensated by increased anaerobic glycolysis to yield pyruvate, which is further converted by lactate dehydrogenase (LDH) to lactate. This reaction sequence is activated under hypoxic conditions and in cases of high energy demand [120,121]. As the LDH-driven conversion of pyruvate to lactate is accompanied by reduction of NADH, the resulting NAD+ stabilizes the cytoplasmic redox status [122,123]. Lactate is secreted from cells together with H+. Once released, lactate is disposed by other cells within the tissue or transported via bloodstream mainly to liver, brain, heart, and working skeletal muscle, where it is taken up and served as fuel [124,125,126] or is used for gluconeogenesis as in the liver [127]. Furthermore, lactate promotes release of Mg2+ from endoplasmic reticulum, favors gene expression by interaction with histones, and inhibits lipolysis via ligand binding to the G-protein GPR81 [128,129].
Lactate is an anti-inflammatory agent. At sites with enhanced lactate levels, monocytes and macrophages are reprogramed into an anti-inflammatory M2-type [56,57]. In human monocytes and macrophages, extracellular lactate shifts the energy metabolism from glycolysis to oxidative phosphorylation. Whereas short-term lactate exposure has limited effects on cytokine production, long-term exposure reprograms immune cells to anti-inflammatory responses [57]. Lactate inhibits monocyte migration and cytokine release [130,131].
When glycolysis prevails for ATP production, mitochondria serve mainly as biosynthetic organelles to yield initial components for synthesis of lipids, nucleotides, and non-essential amino acids [132]. The uptake of glutamine increases markedly under these conditions. The stepwise conversion of glutamine into α-ketoglutarate provides the basis for synthesis of biological molecules independent of glucose metabolism. For example, α-ketoglutarate is converted via the intermediate formation of citrate into acetyl-Co-A, which is necessary for lipogenesis [133].
The aforementioned deviations in energy metabolism in aged individuals affect general physiological processes, induce numerous adaptations to altered cellular metabolic routes, and promote inflammatory reactions by release of DAMPs from defective cells. In particular, oxidative stress and oxidants as cytotoxic agents play an increasing role under these conditions.
3.3. Responses to Hypoxia and Oxidative Stress
In order to better understand the real nature of inflammatory processes in individuals of advanced age, it is necessary to focus on basic mechanisms how cell and tissue homeostasis can be maintained over long time under conditions of increased impacts. In elderly persons, these impacts result predominantly from deficits in dioxygen and nutrient’s supply. Hypoxia is a key factor affecting metabolic routes in elderly persons [134,135,136]. Without considering external reasons for generalized hypoxia such as dioxygen deficiency in the breathing gas (e. g. stay of people at high altitude), at advanced age hypoxic conditions are often the consequence of a diminished blood flow, lung problems, or the reduced capacity of blood cells to carry dioxygen. In addition, health problems and enhanced blood pressure values are other major players in formation of hypoxic conditions.
In the affected tissue regions, hypoxia causes activation and stabilization of cytosolic hypoxia-inducible factor 1α (HIF-1α), which up-regulates glycolysis, down-regulates mitochondrial oxidative phosphorylation, and favors the enhanced formation of reactive species in dysfunctional mitochondria [137,138,139]. In highly vascularized tissues, HIF-1α is replaced by HIF-2α during prolonged hypoxia [140,141].
HIF-1α is a master regulator of glycolysis in muscle and many other cells. Cytosolic HIF-1α is usually tagged by prolyl hydroxylase for proteasome degradation [142,143]. Essential cofactors for active prolyl hydroxylase are O2, Fe2+, and α-ketoglutarate [144]. This proteasomal degradation of HIF-1α is lost under hypoxic conditions due to lack of O2 [137]. In addition, HIF-1α is known to up-regulate transcription of several enzymes promoting glycolysis such as hexokinase II (HK2), pyruvate kinase M2 (PKM2), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase-1 (PDHK1). The latter enzyme is a negative regulator of pyruvate dehydrogenase (PDH) [138]. As a result, pyruvate is mainly metabolized to lactate, but not to acetyl-CoA in hypoxic cells.
In addition to stabilization of cellular redox processes, long-lasting adaptations to hypoxia mainly concern the over-expression of proteins antagonizing oxidative stress. Activation of the stress-sensitive transcription factor Nrf2 protects cells against damaging reactions of reactive species and related oxygen-based cytotoxic agents [145,146,147]. Under stress conditions, superoxide dismutase, catalase, glutathione peroxidase and other protecting antioxidative proteins can be up-regulated. This counter-regulates at least partially or more efficiently stress-related oxidant-based cytotoxic agents. Several potential scenarios determine the further fate of affected cells and tissues. First, cell metabolism can be stabilized by up-regulation of protective antioxidant systems and cells survive as found in many cancers. Second, processes of autophagy and mitophagy are enhanced to remove damaged cell material and defective organelles. Third, last but not least, cells are driven into apoptosis or necrosis. These scenarios have fundamental consequences for the (patho)physiological features of hypoxic areas.
Enhanced expression of hypoxia-induced factors (HIFs) causes numerous gene-induced adaptations to stabilize the cellular redox status, to resist against oxidative stress, and to reprogram cell metabolism. Enhanced intracellular values of reactive species and other oxidant-based cytotoxic agents activate the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) that plays a key role in enhanced synthesis of antioxidant proteins [145,146]. Under normal physiological conditions Nrf2 is complexed with the by Kelch like-ECH-associated protein 1 (KEAP1) and Cullin 3, which cause ubiquitination and degradation of Nrf2 [147]. Cellular stress releases Nrf2 from this complex and promotes nuclear transcriptional processes by Nrf2. Genes activated by Nrf2 code proteins, which are involved in carbohydrate metabolism including NADPH generation, redox regulation and antioxidant defense systems like glutathione and thioredoxin mediated reactions, heme detoxification, iron metabolism, lipid metabolism, proteasomal degradation, autophagy, apoptosis, and biotransformation and detoxification [148].
3.4. Redox Regulation and Antioxidative Defense in Aged Individuals
Many physiological functions of cells depend on the presence of a reducing milieu. A high cytoplasmic level of NADPH is mandatory to ensure redox homeostasis in cells. NADPH is the major electron source within cells [123]. It is oxidized to NADH+ by glutathione reductase [149] and thioredoxin reductase [150,151] keeping both glutathione (GSH) and thioredoxin (Trx) in their reduced states. In quiescent cells, the ratio [NADPH]/[NADP+] is approximately 100:1 [123]. Two enzymes of the pentose phosphate pathway that runs parallel to glycolysis, namely glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase, reduce NADP+ to NADPH [152]. An overview about basic mechanisms of cellular redox homeostasis and potential alterations under stress situations is given in Figure 3. In contrast, in the redox couple NAD+/NADH, the oxidized form predominates giving values between 1:10 and 1:1000 for the ratio [NADH]/[NAD+] [123]. Interconversions between members of both nicotinamide-based redox couples are controlled by NADH kinases and NADPH phosphatases [153]. By these reactions and the presence of numerous cytosolic binding sites for NADH and NADP+ [123] both redox couples are kept far from equilibrium. These conditions allow to fulfil them key physiological functions in maintenance of redox homeostasis.
With advanced age, the increasing significance of glycolysis has several consequences for the redox status. In addition, cytoplasmic and mitochondrial stress increases too. The higher demand of NADPH can be compensated by enhancement of the pentose phosphate pathway. The enhanced formation of lactate from pyruvate is coupled to reduction of NADH to NAD+. The latter species is a cofactor for numerous enzymes including sirtuins, which are important modulators of mitochondrial homeostasis and aging [111,112,113,114]. Both reduced formation of novel NAD+ and higher activity of NAD+ consuming enzymes contribute to age-dependent decline of NAD+ [102,107,108]. Reduced level of NAD+ is associated with defects in oxidative phosphorylation [154,155,156,157,158].
The glutathione system is involved in removal of lipid hydroperoxides, peroxynitrite, and hydrogen peroxide by glutathione peroxidase-mediated reactions, reduction of disulfides, and reduction of glutaredoxin [159]. Glutathione reductase converts oxidized glutathione (GS-SG) to the reduced form (GSH). In addition, GSH can be supplied by de novo synthetic processes. In elderly individuals, disturbance of components of redox metabolism highly depends on the intactness of immune functions. Thus, in this cohort, improved immune functions correlate with higher values of glutathione peroxidase, glutathione reductase, GSH, and GSH/GS-SG ratio [160]. Impaired immune functions are associated with lower levels of these parameters. Investigation of large groups of healthy humans revealed a gradual decline of the GSH content with increasing age in blood and blood cells [161,162,163]. A gradual decrease of GPx activity is reported in female persons after age 65 [164]. In brain of aged persons, impaired glutathione is associated with different neurological disorders [165,166].
The thioredoxin system is another major contributor to redox homeostasis in cells. Reduced thioredoxin (Trx) is not only an essential factor to reduce cellular oxidized proteins such as oxidized peroxiredoxins, ribonucleotide 5’-diphosphatase, and methionine sulfoxide [159]. It exerts also an important regulatory role as cofactor for several enzymes and transcription factors. In other words, Trx is regarded as a biomarker for age-related diseases, oxidative stress, inflammation, and cellular senescence [167]. For example, reduced Trx forms complexes with apoptosis-signal-regulating protein 1 (ASK1) and phosphates and tensin homolog (PTEN) keeping these binding partners in an inactive state [168].
Enhanced cellular damage, redox imbalance, and disease progression are linked to diminished Trx and activation of the thioredoxin-interacting protein (Txnip) [169] and activation of proteins like ASK1 and PTEN, which both are involved in apoptosis induction. In cells, Txnip forms a complex with reduced Trx via formation of a disulphide bond [170]. Oxidative stress as well as Trx deficiency leads to decomposition of this inhibitory complex and allows Txnip to interact with other target molecules. Txnip activates the (NOD)-like receptor protein-3 (NRLP3) inflammasome complex via interaction of Txnip with the proapoptotic protein ASK1 [171,172]. The inflammasome complex contributes to mitochondrial stress-induced apoptosis via expression of inflammatory cytokines. In addition, Txnip binds to phosphatase and tensin homolog (PTEN) and favors thus apoptosis induction through regeneration of PIP2 [168]. Txnip suppresses also glycolysis and promotes oxidative phosphorylation. Glucose transporters 1 and 4 are up-regulated by Txnip [157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173]. The expression of Txnip in blood precursor cells implies a role of Txnip during hematopoiesis [176].
Txnip is up-regulated during aging in primary human cells [177]. This up-regulation is associated with increased oxidative stress, DNA damage, and reduced lifespan [177]. In diabetes patients, Txnip is up-regulated too [178,179,180]. Txnip is also implicated in the pathogenesis of Alzheimer disease and Morbus Parkinson [181,182,183]. In brain samples of patients with Alzheimer disease, less neuronal Txnip was observed in comparison to unperturbed brains. However, an increased number of microglia cells was found in these samples, which were associated with Txnip-positive plaques [184].
Otherwise, Txnip is a negative regulator of cell proliferation. Up-regulated Txnip inhibits cyclin A and several others factors promoting cell cycle [169]. Cancer cell growth is associated with decreased Txnip, increased glycolytic flux, and enhancement of proliferative processes [185,186].
In addition to the aforementioned components of the glutathione and thioredoxin systems, further proteins and enzymes contribute to defense against oxidative stress. In drosophila and some other invertebrate models, over-expression of superoxide dismutase (SOD) has been associated with protection against reactive species and extension of lifespan [187,188]. Otherwise, SOD activity is dispensable for normal animal aging but is useful in stress situations [189]. In humans, a potential relationship between SOD and aging remains unclear. In older women, higher level of plasma SOD activity was linked to lower overall mortality, but not in older men [190]. SODs are essential to catalyze the dismutation of superoxide anion radicals, which mainly result from dysfunctional mitochondria, activated immune cells, and other sources, into dioxygen and hydrogen peroxide. In this way, SODs prevent side reactions of superoxide anion radicals with nitrogen oxide to yield the powerful oxidant peroxynitrite [191,192] and with iron-sulfur clusters of some mitochondrial proteins resulting in release of catalytically active Fe2+ [118,119].
Hydrogen peroxide is inactivated by glutathione peroxidases, catalase, and peroxiredoxins. Catalase malfunction or deficiency is hypothesized to play a role in the pathogenesis of age-related diseases such as diabetes mellitus, hypertension, neurodegenerative disorders, vitiligo, and some others [193]. Age-related alterations in functions of peroxiredoxins are mainly obtained from experiments with mice [194]. Peroxiredoxin 3 (Prx3), the mitochondrial form of peroxiredoxins, is involved in maintenance of mitochondrial redox homeostasis [195,196]. As the expression of Prx3 declines with age mitochondrial functions and homeostasis are impaired [197]. Prx1 contributes to dampen telomere shortening, a key age-dependent process [198,199,200].
Iron is an essential cofactor of many proteins. Within mitochondria, iron is part of many proteins in form of iron-sulfur-clusters and as central ion in the heme group of heme proteins. As free iron ions have a high potential to catalyze damaging reactions in cells, all aspects of iron transport, utilization, and metabolism are usually well controlled [201,202]. In cells, transferrin receptor (iron uptake), ferritin (iron storage), and ferroportin (iron export) are major players to ensure iron homeostasis and utilization [66,67,68,69]. In aged individuals, increased ferritin level is associated with the presence of a chronic inflammatory state [203]. Otherwise, lower ferritin levels are found in patients with iron deficiency [203].
Similar conclusions concerning transport, utilization, and metabolism can be drawn for copper ions. In blood, copper is mostly bound and transported by ceruloplasmin followed by serum albumin [204,205]. The copper-containing ceruloplasmin detoxifies Fe2+ by oxidation to Fe3+ and contributes thus to maintenance of iron homeostasis [206]. A glycosylphosphatidylinositol (GPI)-anchored form of ceruloplasmin [207] is present in astrocytes, oligodendrocytes, hepatocytes, macrophages, and epithelial cells in pancreas and retina [208,209] and is part of the iron efflux machinery [210]. GPI-anchored ceruloplasmin oxidizes intracellular Fe2+ to Fe3+, which is excreted by ferroportin and further safely transferred to transferrin [211]. Patients deficient in ceruloplasmin develop massive iron deposition in brain, liver, and other organs and exhibit neurological symptoms and motor deficits [212,213,214]. Similarly, ceruloplasmin knockout mice are characterized by iron accumulation in astrocytes, ferritin expression, and neurodegeneration in the central nervous system [207,215].
The uncontrolled increase of the free labile pool of iron ions can lead to ferroptosis. In this kind of cells death, enhanced levels of free iron ions catalyze the oxidation of lipid hydroperoxides, which has fatal consequences on the barrier function of biological membranes. These mechanisms are promoted by glutathione deficiency and decreased activity of glutathione peroxidase 4 [216,217]. The latter enzyme is together with its cofactor glutathione responsible for detoxification of lipid hydroperoxides [62]. Another player in removal of lipid hydroperoxides and prevention of ferroptosis is peroxiredoxin 6 [218,219].
In plasma, haptoglobin binds hemoglobin after its release from red blood cells and myoglobin resulting from damaged muscles [220,221]. The haptoglobin-heme protein complex is removed from circulation by spleen and liver macrophages [222,223]. Both released hemoglobin and myoglobin can liberate the powerful oxidant ferriprotoporphyrin IX, also known as free heme [224]. Hemopexin inactivates free heme by forming a high-affinity complex with free heme and clearing this complex in the liver [225]. In elder humans, blood level of haptoglobin is elevated [226]. Another study revealed enhanced serum values for the haptoglobin-hemoglobin complex with age [227]. These data indicate an enhanced leakage of red blood cells and maybe problems with the clearance of the haptoglobin-heme protein complex in aged individuals.
In cell cytoplasm, hemoxygenase 1 (HO-1) degrades free heme to biliverdin, carbon monoxide, and Fe2+. The role of HO-1-derived Fe2+ is controversially discussed [228,229]. Low amounts of Fe2+ can be stored in ferritin and induce synthesis of additional ferritin [230]. Higher levels of Fe2+ and overload of ferritin with iron can promote ferroptosis [231]. There is a decline of HO-1 expression with increasing age in macular and peripheral retinal pigment epithelium [229,232].
3.5. Activation of Proteolytic Systems in Aged Individuals
Normally, processes of protein synthesis and degradation are in equilibrium in living tissues. With increasing age, however, degradative mechanisms prevail. This is mainly caused by disturbances in energy metabolism, dioxygen and nutrient deficiencies, and increased defects in cellular constituents and organelles in association with stress situations.
There are several levels how cell organelles and cells respond to stress situations. Mitochondrial unfolded protein response (UPR) is activated when the number of unfolded proteins is increased beyond a certain threshold. In this pathway, genes are activated favoring the translation of proteins involved in detoxification of reactive species, supporting the correct folding, and removing misfolded proteins [102]. In mouse models, positive effects of enhanced levels of NAD+ have been demonstrated on improvement of UPR and mitochondrial homeostasis [154,233]. As UPR links mitochondrial and nuclear genomes, dysregulation of this pathway is reported in several age-related human diseases like sarcopenia, and Morbus Alzheimer [234,235,236,237].
In stress situations, processes of mitochondrial membrane dynamics, known as mitochondrial fusion and fission, allow to dilute and segregate damaged organelles in order to ensure homeostasis and to survive [238,239]. The dynamin-related protein 1 plays an important role in fission regulation [102]. In mouse models, reduced activity of this protein is impaired with increasing age [240,241].
In cells, defective proteins are recognized and degraded by the ubiquitin-proteasome system [242,243]. With increasing age misfolded and damaged proteins accumulate [244]. During aging induction of chaperones is impaired [245] and proteasomal activity is declining as evidenced in different cells and tissues [246,247,248]. For example, age-related decreased activities of 20S and 26S proteasome complexes are reported [249,250,251].
Damaged cell organelles, dysfunctional cytoplasmic and structural components, protein aggregates, and sometimes invading pathogens are preferentially cleared by autophagic mechanisms by means of lysosomes [252,253]. The selective removal of damaged mitochondria by autophagy is known as mitophagy. Both ubiquitin-dependent and -independent pathways of mitophagy are differentiated [102]. In mice and humans, a decline of mitophagy has been observed in several tissues upon aging [254,255,256,257,258].
Defective mitochondria are linked to immune activation via the release of DAMPs. Putative mitochondrial DAMPs are mitochondrial DNA, cardiolipin, and N-formyl peptides resulting from translation of mitochondrial-encoded proteins [259]. There is a gradual increase of mitochondrial DNA in blood in elderly persons [260]. Mitochondria-derived DAMPs are involved in NLRP3 inflammasome activation, and consequently in release of pro-inflammatory cytokines like IL-1β and IL-18 and apoptosis induction [261,262,263].
Both the ubiquitin-proteasome system and autophagy are intensified in cachectic patients [264]. Cachexia is associated with progressive skeletal muscle wasting and substantial weight loss as a consequence of an underlying illness and is an important comorbidity in cancer patients [265,266,267] as well as in individuals of advanced age [268,269].
3.6. Inflammaging and Necrotic Cell Death
Inflammaging is characterized by increasing number of factors secreted from senescent cells. These factors are summarized as senescence-associated secretory cell phenotype. They include pro-inflammatory cytokines, chemokines, growth factors, proteases, extracellular matrix components and some others [270]. Pathophysiological consequences of inflammaging are very complex. On the cellular level, increased oxidative stress is assumed to be a crucial factor for accumulation of numerous structural and functional defects and initiation of novel inflammatory events [271,272,273]. This interplay between oxidative stress and inflammation during aging is also associated with the increased death of dysfunctional cells. In addition to apoptotic cell death, which occurs usually without further disturbances to neighbored cells and tissues, different forms of necrosis increase with age. These forms include ferroptosis, necroptosis, pyroptosis, and others [270]. During necrotic cell death, both cytotoxic agents as well as different DAMPs are released. These agents contribute to further tissue damage and trigger novel inflammatory events.
A few examples will underline the pathophysiological significance of necrotic cell death in aged persons. In aged liver, increase of necroptosis, a regulated form of programmed necrosis, contributes to chronic liver inflammation and development of liver cirrhosis [274]. The risk for pyroptosis increases in aged cells, as these cells are more susceptible to external stimuli that can cause cell damage [275]. Cytokine release and inflammasome activation by pyroptosis has been linked to development of atherosclerosis and neurodegenerative diseases [276,277]. Iron-dependent lipid peroxidation leads to ferroptotic cell death. This mechanism is discussed to contribute to pathogenesis of neurodegenerative diseases, autoimmune disorders, and cardiovascular diseases [278,279].
Increasing immunosuppression is another key feature of inflammaging. During resolution of inflammation, a transient immunosuppression allows to down-regulate all inflammatory processes including apoptosis of immune cells, to induce de novo formation of matrix and tissue components, and to restore the normal tissue homeostasis [43,44]. Under chronic conditions as given in inflammaging, immunosuppressive states can be pronounced more stronger and takes longer. Both enhanced expression of host-derived cytotoxic agents and decline, exhaustion, or inactivation of the corresponding antagonizing principles favor the chronic inflammatory process [8]. Under these conditions, different comorbidities are developing in persons of advanced age. Moreover, long-lasting immunosuppression dampens general immune functions and favors infections with commensal and mutualistic pathogens [280,281,282,283,284]. In the worst case, organ failure, sepsis and septic shock are developing as may occur in immunocompromised individuals [285,286].
4. Age-Related Alterations in the Protection Against Oxidant-Based Cytotoxic Agents in Selected Disease Scenarios
4.1. Diseases of the Cardiovascular System
Disturbances in blood vessel and heart function are very common in elder persons. Most of all, physiological functions in aged individuals are affected by increased blood pressure, hypoxia, alterations in blood vessel elasticity, processes of intravascular hemolysis, internal hemorrhages, and inflammatory processes in vessel walls. Common health problems are atherosclerotic vascular diseases including coronary artery disease, myocardial infarction, cerebrovascular disease, and stroke. Further serious blood flow disturbances are caused by tissue damage due to ischemic and reperfusion events, thrombosis, reduced wound healing, and vasculitis.
Accumulation of cholesterol-rich low-density lipoproteins in damaged endothelium is a hallmark of atherosclerosis. Lipid oxidation, plaque formation, attraction of immune cells, alterations of endothelial cells, and development of foam cells are further characteristics of this disease [287,288,289,290,291,292]. As cholesterol crystals and numerous DAMPs are present in the plaques, novel inflammatory cascades are continuously activated contributing to the chronic inflammatory process.
Increasing age is considered as a risk factor for the development of atherosclerotic vascular diseases [293,294,295,296]. Molecular details of the pathogenesis of atherosclerosis are not fully understood. Nevertheless, it is evident that oxidative processes play an important role in disease progression. In addition to cholesterol and lipid oxidation, the involvement of myeloperoxidase, a heme protein released form recruited neutrophils, is discussed in plaque formation. This enzyme is found in atherosclerotic plaques [297,298], produces the powerful oxidant hypochlorous acid and catalyzes numerous other oxidative processes [299]. The potential participation of free heme presents another route discussed in atherosclerotic plaque formation [300]. As free heme is known to easily penetrate into hydrophobic areas of proteins, membranes, and lipoproteins, it contributes to endothelial damage and lipoprotein oxidation during atherosclerosis [300,301].
Production of nitric oxide (NO) by NO synthases in the vessel wall is crucial for vessel relaxation and regulation of the blood flow. Decreased bioavailability of NO is observed by enhanced processes of intravascular hemolysis from red blood cells or rhabdomyolysis from damaged muscles [221,302]. Both released hemoglobin and myoglobin are known to be rapidly oxidized by NO [303,304,305]. In addition, myeloperoxidase, which can be attached to the inflamed endothelium, interacts with NO and diminishes thus the bioavailability of NO [306].
Another complication in cardiovascular diseases is the formation of internal hemorrhages. These bleedings are associated with uncontrolled hypertension, problems in blood clotting, traumata, aneurysms, loss of blood vessel elasticity, reduced arterial compliance with increasing age, and weakening of collagen linkages to the vessel wall [307,308,309]. In newly formed hemorrhages, there is a loss of glucose, dioxygen, and other nutrients with time. Concomitantly, intravascular hemolysis of red blood cells increases and protecting haptoglobin and hemopexin are exhausting. The resulting cytotoxic free heme can induce numerous damaging reactions at adjacent tissue areas [301,310,311].
4.2. Diabetes Mellitus
Majority of people over 65 years have prediabetes or diabetes, whereby diabetes type 2 predominates [315]. Diabetes is associated with many serious disease complications like vascular problems, neuropathy, nephropathy, and retinopathy [316]. Hyperglycemia leads to dysfunctions of red blood cells, endothelial cells, and other cells. These cells are known to accumulate glucose in an uncontrolled fashion independent of the presence of insulin. As a result, glycation of proteins increases in these cells beyond the level found in healthy individuals and different advanced glycation end products accumulate [317,318].
In red blood cells, glycation of hemoglobin as measured by the HbA1c value serves as a long-term marker for hyperglycemia [319,320]. In these cells, part of glucose is converted into sorbitol, which cannot leave the cells [321]. Accumulated sorbitol contributes to increased stiffness and diminished deformability of red blood cells [322,323,324]. Hence, these cells become osmotically instable and intravascular hemolysis increases [323]. Vascular endothelial cells develop an inflammatory phenotype on hyperglycemia [325,326]. These alterations diminish the blood flow by reduced bioavailability of nitric oxide, decreased vasorelaxation, impaired fibrinolytic activities, changes in the cellular cytoskeleton, and thickening of the basal lamina [326,327,328]. In addition, release of the von Willebrand factor from endothelial cells exposed to high levels of glucose can initiate thrombolytic events and adverse cardiovascular complications [329,330,331].
4.3. Cancer
With advanced age, the incidence increases for the development of different kinds of cancers. Which concrete conditions contribute to cancer development in a given patient remains puzzling. Key factors for cancer development in aged individuals are the increasing hypoxia in tissues and underlying problems in immune defense due to the presence of a low-stage chronic inflammation.
In patients with advanced cancers, host-own protective mechanisms are triggered in a way that they support cancer growth and metastasis formation rather than recognition and elimination of cancer cells by the immune system [332,333]. In cancer cells, antioxidative proteins and mechanisms stabilizing cellular redox homeostasis are highly up-regulated [9]. By these adaptations, cancer cells tolerate enhanced values of reactive species. This allows them also to survive even therapeutic approaches like radio- and chemotherapy. Moreover, in the tumor microenvironment (TME), invading immune cells are in a permanent state of immunosuppression [334,335,336,337] that correspond to down-regulated immune functions during the resolution phase of an acute inflammation [9]. Cancer cells release several factors such as tumor-necrosis factor β, IL-10, vascular endothelial growth factor, and components contained in secreted exosomes, which promote immunosuppression and support resolution of inflammation [338].
High up-regulation of glycolysis with lactate as final product is a metabolic hallmark in cancer cells. Anaerobic glycolysis supplies the necessary energy equivalents in these cells instead of the dioxygen-dependent mitochondrial oxidative phosphorylation. Secreted lactate can be used in the TME as fuel by invading macrophages to trigger them into an anti-inflammatory subtype M2 [56,339]. Enhanced expression of carbonic anhydrases on the external surface of cancer cells and up-regulation of several transport proteins in the plasma membrane acidifies the pH value in TME and decreases the cytoplasmic proton concentration [340,341,342,343]. The resulting mild alkalization in cancer cell cytosol is typical of proliferating cells. In other words, enhancement of glycolysis and subsequent metabolic alterations support cancer growth, metastasis, and invasion [344,345].
4.4. Neuro-Degenerative Diseases
Neuronal plaque deposits determine the morphological picture of neuro-degenerative diseases like Alzheimer’s disease, Parkinson’s disease, and others. These diseases often appear in persons of advanced age. The accumulation of fibrillated proteins coincides with a progressive loss of mental abilities due to the loss of functional neurons and synaptic linkages.
In Alzheimer’s disease, large extracellular deposits of amyloid β and hyperphosphorylated τ protein accumulates [346,347]. However, molecular details supporting fibrillation are largely unknown. Disturbances in endosomal-lysosomal and autophagic pathways contribute to processing of peptide fragments into insoluble fibrils. During fibril formation, soluble proteins with dominating α-helices are converted into insoluble proteins with preferred β-sheets [348,349]. This conversion is favored by acidic pH values as found in endosomes. Pathogenesis of Alzheimer’s disease is linked to the presence of very low pH values in endosomes [350]. This hyper-acidification enhances the formation of amyloid β fibrils and affects also the clearance of these fibrils.
Postmortem measurements revealed a slight decrease of pH values in the brain and cerebrospinal fluid of humans and mice with increasing age [351]. In Alzheimer’s patients, pH values of brain and cerebrospinal fluid were lower compared to controls without this pathology [351,352]. Moreover, lower brain pH values were associated with a more severe course of the disease [352].
Disturbances in the iron metabolism are also discussed in the pathogenesis of neuro-degenerative disorders. Enhanced values of brain iron are observed in several disorders named neurodegeneration with brain iron accumulation [353]. Iron accumulation within mitochondria contributes to oxidative stress, mitochondrial dysfunction, and neurodegeneration in Alzheimer’s disease, Parkinson’s disease and others [354]. Unbalanced iron ions are able to oxidize lipids and other cell constituents that leads in consequence to cell death by ferroptosis [355].
In Morbus Wilson, excessive accumulation of copper ions in liver, brain, and other organs is associated with serious damaging reactions [356]. In Alzheimer’s disease, increased values of free copper have been observed, which are not bound to ceruloplasmin [357,358].
Dysfunctions in heme metabolism are another source for neurotoxic events. In intracerebral and subarachnoid hemorrhages, the powerful oxidant free heme can result from released hemoglobin of defective red blood cells [359,360] after exhaustion of the protecting proteins haptoglobin and hemopexin [222,223,225]. The flat and hydrophobic free heme inserts easily into biological membranes, lipoproteins and hydrophobic areas of proteins. At these loci, it favors oxidative processes and contributes thus to development of atherosclerotic lesions [300], lipid peroxidation [301], neurodegenerative processes [360], and induction of inflammatory events [32,361]. Amyloid β forms a complex with free heme, which promotes the formation of amyloid β fibrils [362]. Enhanced levels of haptoglobin in serum of Alzheimer’s patients versus control support the hypothesis that neuroinflammation and heme-induced stress contribute to pathogenesis of Alzheimer’s disease [363]. Contrariwise, hemopexin level is lower in the cerebrospinal fluid in patients with Alzheimer’s disease [364]. The different behavior between haptoglobin and hemopexin is maybe caused by the fact that human haptoglobin is an acute phase protein unlike human hemopexin [365,366].
5. Conclusions
Our organism is equipped with powerful protective systems to resist a wide variety of external and internal impacts, to repair any damage, and to restore and maintain cell and tissue homeostasis. In many physiological processes, during activation of immune cells, and under stress situations, an enhanced formation of host-derived cytotoxic agents can occur. To avoid any damage by these agents, ready-to-use antagonizing principles exist, which immediately inactivate and eliminate these agents. Serious problems arise when cytotoxic agents are very strong expressed and act over a long time. Under these conditions, protection is limited and cells and tissues are progressively damaged.
During aging, there is a gradual decline in production of energy equivalents, and supply of tissues with dioxygen and nutrients. On this background, a chronic inflammatory state is developing in aged persons. In this persistent inflammation, known as inflammaging, activated immune cells, and pro-inflammatory mediators are present and coincides with a state of immunosuppression. In turn, oxidant-based cytotoxic agents affect considerably the cell and tissue integrity in aged individuals and contribute to the development of age-related diseases. The balance between cytotoxic agents and antagonizing principles is crucial for the further fate of an inflammation. In stressed and hypoxic tissues, there is an up-regulation of components of cellular redox homeostasis and anti-oxidative proteins. These adaptations have, however, long-lasting consequences for the distribution of energy substrates in relation to other physiological processes. More and more energy substrates are needed to stabilize protective mechanisms. These energy substrates are lacking for other physiological processes like active movement, mental processes, sensing the environment, and creating reserves for immunological defense.
It is very hard to predict, how the imbalance between cytotoxic agents and protective mechanisms will affect the health status in a given person during aging. As protection against different oxidants and other cytotoxic agents plays an important role in stabilizing immune responses and preventing disease progression, a thorough analysis of the status of protective mechanisms is highly mandatory for aged individuals.
Conflicts of Interest
The author declares no conflict of interests.
References
- Medvedev, Z.A. An attempt at a rational classification of theories of aging. Biol. Rev. 1990, 375-398. [CrossRef]
- Arnhold, J. Aging in complex multicellular organisms. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 231–247. [CrossRef]
- Arnhold, J. Cells and organisms as open systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 3–22. [CrossRef]
- Arnhold, J. Immune response and tissue damage. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 155–204. [CrossRef]
- Arnhold, J. Acute-phase proteins and additional protective systems. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 205–228. [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; de Luca, M.; Ottaviani, E.; de Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 244-254. [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505-522. [CrossRef]
- Arnhold, J. Host-derived cytotoxic agents in chronic inflammation and disease progression. Int. J. Mol. Sci. 2023, 24, 3016. [CrossRef]
- Arnhold, J. Inflammation-associated cytotoxic agents in tumorigenesis. Cancers 2024, 16, 81. [CrossRef]
- Franceschi, C. Cell proliferation, cell death and aging. Aging Clin. Exp. Res. 1989, 1, 3-15. [CrossRef]
- Kirkwood, T.B.L.; Franceschi, C. Is ageing a complex as it would appear? New perspectives in gerontological research. Ann. N. Y. Acad. Sci. 1992, 663, 412-417. [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; Cevenini, E.; Castellani, G.C.; Salvioli, S. Inflammaging and antiinflammaging: A systemic perspective on aging and longevity emerged from studies on humans. Mech. Aging Dev. 2007, 128, 92-105. [CrossRef]
- Baggio, G.; Donazzan, S.; Monti, D. Mari, D.; Martini, S.; Cabelli, C.; Dalla Vestra, M.; Previato, L.; Guido, M.; Pigozzo, S.; Cortella, I.; Grepaldi, G.; Franceschi, C. Lipoprotein(a) and lipoprotein profile in healthy centenarians: A reappraisal of vascular risk factors. FASEB J. 1998, 12, 433-437. [CrossRef]
- Gangemi, S.; Basile, G.; Merendino, R.A.; Minciullo, P.L.; Novick, D.; Rubinstein, M.; Dinarello, C.A.; Lo Balbo, C.; Franceschi, C.; Basili, S.; D’Urbano, E.; Davi, G.; Nicita-Mauro, V.; Romano, M. Increased circulating interleukin-18 levels in centenarians with no signs of vascular disease: Another paradox of longevity. Exp. Gerontol. 2003, 38, 669-672. [CrossRef]
- Mannucchi, P.M.; Mari, D.; Merati, G.; Peyvandi, F.; Tagliabue, L.; Sacchi, E.; Taioli, E.; Sansoni, P.; Bertolini, S.; Franceschi, C. Gene polymorphism predicting high plasma levels of coagulation and fibrinolysis proteins. A study in centenarians. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 755-759. [CrossRef]
- Coppola, R.; Mari, D.; Lattuada, A.; Franceschi, C. Von Willebrand factor in Italian centenarians. Haematologica 2003, 88, 39-43.
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-related diseases. J. Gerontol. Biol. Sci. 2014, S4-S9. [CrossRef]
- Troiano, L.; Pini, G.; Petruzzi, E.; Ognibene, A.; Franceschi, C.; Monti, D.; Casotti, G.; Cilotti, A.; Forti, G. Evaluation of adrenal function in aging. J. Endocrinol. Invest. 1999, 22, 74-75.
- Carrieri, G.; Marzi, E.; Olivieri, F.; Marchigiani, F.; Cavallone, L.; Cardelli, M.; Giovanetti, S.; Stecconi, R.; Molendini, C.; Trapassi, C.; De Benedictis, G.; Kletsas, T.; Franceschi, C. The G/C915 polymorphism of transforming growth factor beta1 is associated with human longevity: A study in Italian centenarians. Aging Cell 2004, 3, 443-448. [CrossRef]
- Baylis, D.; Bartlett, D.B.; Patel, H.P.; Roberts, H.C. Understanding how we age: Insights into inflammaging. Longevity Healthspan 2013, 2, 8. [CrossRef]
- Newman, A.B.; Sanders, J.L.; Kizer, J.R.; Boudreau, R.M.; Odden, M.C.; Zeki al Hazzouri, A.; Arnold, A.M. Trajectories of functions and biomarkers with age: The CHS all stars study. Int. J. Epidemiol. 2016, 1135-1145. [CrossRef]
- Xia, S.; Zhang, X.; Zheng, S.; Khanabdali, S.; Kalionis, B.; Wu, J.; Wan, W.; Tai, X. An update of inflamm-aging: Mechanisms, prevention, and treatment. J. Immunol. Res. 2016, 8426874. [CrossRef]
- Dugan, B.; Conway, J.; Duggal, N.A. Inflammaging as a target for healthy ageing. Age Ageing 2023, 52, 1-15. [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transd. Target. Ther. 2023, 8, 329. [CrossRef]
- Suresh, R.; Moser, D.M. Pattern recognition in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells trigger inflammation. Nature 2002, 418, 191-195. [CrossRef]
- Shi, Y.M.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516-521. [CrossRef]
- Scheibner, K.A.; Lutz, M.A.; Boodoo, S.; Fenton, M.J.; Powell, J.D.; Horton, M.R. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 2006, 177, 1272-1281. [CrossRef]
- Bours, M.J.; Swennon, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 2006, 112, 358-404. [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliviera, M.F.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221-20229. [CrossRef]
- Farkas, A.M.; Kilgore, T.M.; Lotze, M.T. Detecting DNA: Getting and begetting cancer. Curr. Opin. Investig. Drugs 2007, 8, 981-986.
- Pepys, M.B.; Baltz, M.I. Acute phase proteins with special reference to C-reactive protein and related proteins (pentraxins) and serum amyloid A protein. Adv. Immunol. 1983, 34, 141–212. [CrossRef]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the death: The impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 2006, 129, 1673–1682. [CrossRef]
- Pober, J.S.; Sessa, W.C. Inflammation and the blood microvascular system. Cold Spring Harbor Perspect. Biol. 2015, 7, 016345. [CrossRef]
- Young, B.; Gleeson, M.; Cripps, A.W. C-reactive protein. A critical review. Pathology 1991, 23, 118-124. [CrossRef]
- Gewurz, H.; Mold, C.; Siegel, J.; Fiedel, B. C-reactive protein and acute phase response. Adv. Intern. Med. 1982, 27, 345-372.
- Kopf, H.; de la Rosa, G.M.; Horward, O.M.; Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 2007, 7, 1819-1824. [CrossRef]
- Rodriguez-Prados, J.C.; Través, P.C.; Cuenca, J.; Rico, D.; Aragonés, J.; Martin-Sanz, P.; Casante, M.; Boscá, L. Substrate fate in activated macrophages; a comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605-614. [CrossRef]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blaigh, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; Pearce, E.J. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742-4749. [CrossRef]
- Morrison, T.; Watts, E.R.; Sadiku, P.; Walmsley, S.R. The emerging role for metabolism in fueling neutrophilic inflammation. Immunol. Rev. 2023, 314, 427-441. [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune response. Annu. Rev. Immunol. 2006, 24, 99–146. [CrossRef]
- Li, M.O.; Flavell, R.A. Contextual regulation of inflammation: A duet of transforming growth factor-beta and interleukin-10. Immunity 2008, 28, 468–476. [CrossRef]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [CrossRef]
- Landén, N.X.; Li, D.; Ståhle, M. Transition from inflammation to proliferation: A critical step during wound healing. Cell Mol. Life Sci. 2016, 73, 3861–3885. [CrossRef]
- Marega, M.; Chen, C.; Bellusci, S. Cross-talk between inflammation and fibroblast growth factor 10 during organogenesis and pathogenesis: Lessons learnt from the lung and other organs. Front. Cell Dev. Biol. 2021, 9, 656883. [CrossRef]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [CrossRef]
- Chandrasekharan, J.A.; Sharma-Walia, N. Lipoxins: nature’s way to resolve inflammation. J. Inflamm. Res. 2015, 8, 181–192. [CrossRef]
- Sanchez-Pino, M.D.; Dean, M.J.; Ochoa, A.C. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 2021, 362, 104302. [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Montavani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [CrossRef]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [CrossRef]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; Zhang, J.; Wang, C.; Shen, J.; Che, Y.; Liu, Z.; Lv; Y.; Wen, H.; You, Q. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428-438. [CrossRef]
- Ratter, J.M.; Rooljackers, H.M.M.; Hoolveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. In vitro and in vivo effects of lactate on metabolism and cytokine production of human primary PBMCs and monocytes. Front. Immunol. 2018, 9, 2564. [CrossRef]
- McCord, J.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1960, 224, 6049–6055.
- Chang, L.Y.; Slot, J.W.; Geuze, H.J.; Crapo, J.D. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. J. Cell Biol. 1988, 107, 2169–2179. [CrossRef]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide dismutase. Site of synthesis and intramolecular localization. J. Biol. Chem. 1973, 248, 4793–4796.
- Antonyuk, S.V.; Strange, R.W.; Marklund, S.L.; Hasnain, S.S. The structure of human extracellular copper-zinc superoxide dismutase at 1.7 Å resolution: Insights into heparin and collagen binding. J. Mol. Biol. 2009, 388, 310–326. [CrossRef]
- Ursini, F.; Maiorino, M.; Roveri, A. Phospholipid hydroperoxide glutathione peroxidase (PHGPx): More than an antioxidant enzyme? Biomed. Environm. Sci. 1997, 10, 327–332.
- Low, F.M.; Hampton, M.P.; Winterbourn, C.C. Prx2 and peroxide metabolism in the erythrocyte. Antioxid. Redox Signal. 2008, 10, 1621–1630. [CrossRef]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protect. Cell 2010, 1, 888–897. [CrossRef]
- Gutteridge, J.M.C. Ferrous-salt-promoted damage to deoxyribose and benzoate. The increased effectiveness of hydroxyl-radical scavengers in the presence of EDTA. Biochem. J. 1987, 18, 37-49. [CrossRef]
- Zhao, N.; Zhang, A.-S.; Enns, C.A. Iron regulation by hepcidin. J. Clin. Investig. 2013, 123, 2337–2343. [CrossRef]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 2011, 1820, 188–202. [CrossRef]
- Gamella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [CrossRef]
- Massover, W.H. Ultrastructure of ferritin and apoferritin: A review. Micron 1993, 24, 389–437. [CrossRef]
- Prohaska, J.R. Role of copper transporters in copper homeostasis. Am. J. Clin. Nutr. 2008, 88, 826S–829S. [CrossRef]
- Sokolov, A.V.; Ageeva, K.V.; Pulina, M.O.; Cherkalina, O.S.; Samygina, V.R.; Vlasova, I.I.; Panasenko, O.M.; Zakharova, E.T.; Vasilyev, V.B. Ceruloplasmin and myeloperoxidase in complex affect the enzymatic properties of each other. Free Radic. Res. 2008, 42, 989–998. [CrossRef]
- Chapman, A.L.P.; Mocatta, T.J.; Shiva, S.; Seidel, A.; Chen, B.; Khalilova, I.; Paumann-Page, M.E.; Jameson, G.N.L.; Winterbourn, C.C.; Kettle, A.J. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J. Biol. Chem. 2013, 288, 6464–6477. [CrossRef]
- Samygina, V.R.; Sokolov, A.V.; Bourenkov, G.; Petoukhov, M.V.; Pulina, M.O.; Zakharova, E.T.; Vasilyev, V.B.; Bartunik, H.; Svergun, D.I. Ceruloplasmin: Macromolecular assemblies with iron-containing acute phase proteins. PLoS ONE 2013, 8, e67145. [CrossRef]
- Sokolov, A.V.; Kostevich, V.A.; Zakharova, E.T.; Samygina, V.R.; Panasenko, O.M.; Vasilyev, V.B. Interaction of ceruloplasmin with eosinophil peroxidase as compared to its interplay with myeloperoxidase: Reciprocal effect on enzymatic properties. Free Radic. Res. 2015, 49, 800–811. [CrossRef]
- Ashby, M.T.; Carlson, A.C.; Scott, M.J. Redox buffering of hypochlorous acid by thiocyanate in physiologic fluids. J. Am. Chem. Soc. 2004, 126, 15976–15977. [CrossRef]
- Nagy, P.; Beal, J.L.; Ashby, M.T. Thiocyanate is an efficient endogenous scavenger of the phagocytic killing agent hypobromous acid. Chem. Res. Toxicol. 2006, 19, 587–593. [CrossRef]
- Davies, M.J.; Hawkins, C.L. The role of myeloperoxidase in biomolecule modification, chronic inflammation, and disease. Antioxid. Redox Signal. 2020, 32, 957–981. [CrossRef]
- Love, D.T.; Barrett, D.J.; White, M.Y.; Cordwell, S.J.; Davies, M.J.; Hawkins, C.L. Cellular targets of the myeloperoxidase-derived oxidant hypothiocyanous acid (HOSCN) and its role in the inhibition of glycolysis in macrophages. Free Radic. Biol. Med. 2016, 94, 88–98. [CrossRef]
- Frommherz, K.J.; Faller, B.; Bieth, J.G. Heparin strongly decreases the rate of inhibition of neutrophil elastase by α1-proteinase inhibitor. J. Biol. Chem. 1991, 266, 15356–15362. [CrossRef]
- Ermolieff, J.; Boudier, C.; Laine, A.; Meyer, B.; Bieth, J.G. Heparin protects cathepsin G against inhibition by protein proteinase inhibitors. J. Biol. Chem. 1994, 269, 29502–29508. [CrossRef]
- Thompson, R.C.; Ohlsson, K. Isolation, properties, and complete amino acid sequence of human secretory leukocyte protease inhibitor, a potent inhibitor of leukocyte elastase. Proc. Natl. Acad. Sci. USA 1986, 83, 6692–6696. [CrossRef]
- Williams, S.E.; Brown, T.I.; Roghanian, A.; Sallenave, J.M. SLPI and elafin: One glove, many fingers. Clin. Sci. 2006, 110, 21–35. [CrossRef]
- Verrier, T.; Solhonne, B.; Sallenave, J.M.; Garcia-Verdugo, I. The WAP protein Trappin-2/Elafin: A handyman in the regulation of inflammatory and immune responses. Int. J. Biochem. Cell Biol. 2012, 44, 1377–1380. [CrossRef]
- Duranton, J.; Adam, C.; Blieth, J.G. Kinetic mechanism of the inhibition of cathepsin G by α1-antichymotrypsin and α1-proteinase inhibitor. Biochemistry 1997, 37, 11239–11245. [CrossRef]
- Travis, J.; Bowen, J.; Baugh, R. Human α1-antichymotrypsin: Interaction with chymotrypsin-like proteinases. Biochemistry 1978, 26, 5651–5656. [CrossRef]
- Kalsheker, N.A. α1-Antichymotrypsin. Int. J. Biochem. Cell Biol. 1996, 28, 961–964. [CrossRef]
- Gordon, J.L. Extracellular ATP: Effects, sources and fate. Biochem. J. 1986, 233, 309-319. [CrossRef]
- Khakh, B.S.; Burnstock, G. The double life of ATP. Sci. Am. 2009, 301, 84-90. [CrossRef]
- Dou, L.; Chen, Y.F.; Cowan, P.J.; Chen, X.-P. Extracellular ATP signaling and clinical relevance. Clin. Immunol. 2018, 188, 67-73. [CrossRef]
- Flood, D.; Lee, E.S.; Taylor, C.T. Intracellular energy production and distribution in hypoxia. J. Biol. Chem. 2023, 299, 105103. [CrossRef]
- El Bacha, T.; Luz, M.R.M.P.; Da Poian, A.T. Dynamic adaptation of nutrient utilization in humans. Nat. Educ. 2010, 3, 8.
- Chandel, N.S. Amino acid metabolism. Cold Spring Harbor Persp. Biol. 2021, 13, a040584. [CrossRef]
- Delgoffe, G.M.; Polizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao. B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295-303. [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell mechanism drives immunity. J. Exp. Med. 2015, 212, 1345-1360. [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91,1447-1531. [CrossRef]
- Ørtenblad, N.; Westerblad, H.; Nielsen, J. Muscle glycogen stores and fatigue. J. Physiol. 2013, 591, 4405-4413. [CrossRef]
- Jensen, T.E.; Richter, E.A. regulation of glucose and glycogen metabolism during and after exercise. J. Physiol. 2012, 590, 1069-1076. [CrossRef]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412-416.
- Holeček, M. Origin and roles of alanine and glutamine in gluconeogenesis in the liver, kidneys, and small intestine under physiological and pathological conditions. Int. J. Mol. Sci. 2024, 25, 7037. [CrossRef]
- Bratic, I.; Trifunovic, A. Mitochondrial energy metabolism and aging. Biochim. Biophys. Acta. 2010, 1707, 961-967. [CrossRef]
- Santanasto, A.J.; Glynn, N.W.; Jubrias, S.A.; Conley, K.E.; Boudreau, R.M.; Amati, F.; Mackey, D.C.; Simonsick, E.M.; Strotmeyer, E.S.; Coen, P.M.; Goodpaster, B.H., Newman, A.B. Skeletal muscle mitochondrial function and fatigability in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 1379-1385. [CrossRef]
- Lima, T.; Li, T.Y.; Mottis, A.; Auwerx. Pleiotropic effects of mitochondria on aging. Nat. Aging 2022, 2, 199-213. [CrossRef]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E. Jr. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575-591. [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 238463. [CrossRef]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A 2010, 65, 119-128. [CrossRef]
- Marzetti, E.; Leeuwenburgh, C. Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 2006, 41, 1234-1238. [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.-S.; Viswanathan, M.; Schoonjans, K.; Guarente, L.; Auwerx, J. The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 2013, 154, 430–441. [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.Y.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; T eodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; Mercken, E.M.; Palmeira, C.M.; de Cabo, R.; Rolo, A.P.; Turner, N.; Bell, E.L.; Sinclair, D.A. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [CrossRef]
- Harrison, A.P.; Pierzynowski, S.G. Biological effects of 2-oxoglutarate with particular emphasis on the regulation of protein, mineral and lipid absorption/metabolism, muscle performance, kidney function, bone formation and cancerogenesis, all viewed from a healthy ageing perspective state of the art–review article. J. Physiol. Pharmacol. 2008, 59, Suppl. 1, 91–106.
- Tian, Q.; Zhao, J.; Yang, Q.; Wang, B.; Deavila, J.M.; Zhu, M.-J.; Du, M. Dietary α-ketoglutarate promotes beige adipogenesis and prevents obesity in middle-aged mice. Aging Cell 2020, 19, e13059. [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [CrossRef]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD+ homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic potential of NAD-boosting molecules: The in vivo evidence. Cell Metab. 2018, 27, 529–547. [CrossRef]
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. npj Aging Mech. Dis. 2016, 2, 16017. [CrossRef]
- Fasolino, M.; Liu, S.; Wang, Y.; Zhou, Z. Distinct cellular and molecular environments support aging-related DNA methylation changes in the substantia nigra. Epigenomics 2017, 9, 21–31. [CrossRef]
- Wang, Y.; Deng, P.; Liu, Y.; Wu, Y.; Chen, Y.; Guo, Y.; Zhang, S.; Zheng, X.; Zhou, L.; Liu, W.; Li, Q.; Lin, W.; Qi, X.; Ou, G.; Wang, C.; Yuan, Q. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat. Commun. 2020, 11, 5596. [CrossRef]
- Borkum, J.M. The tricarboxylic acid cycle as a central regulator of the rate of aging: Implications for metabolic interventions. Adv. Biol. 2023, 7, 2300095. [CrossRef]
- Gardner, P.R. Superoxide-driven aconitase Fe-S cycling. Biosci. Rep. 1997, 17, 33–42. [CrossRef]
- Gardner, P.R. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol. 2002, 349, 9–23. [CrossRef]
- Harmer, A.R.; Chisholm, D.J.; McKenna, M.J.; Hunter, S.K.; Ruell, P.A.; Naylor, J.M.; Maxwell, L.J.; Flack, J.R. Sprint training increases muscle oxidative metabolism during high-intensity exercise in patients with type 1 diabetes. Diabetes Care 2008, 31, 2097–2102. [CrossRef]
- Levy, B.; Gibot, S.; Franck, P.; Cravoisy, A.; Bollaert, P.-E. Relation between muscle Na+K+ATPase activity and raised lactate concentrations in septic shock: A prospective study. Lancet 2005, 365, 871–875. [CrossRef]
- Nalbandian, M.; Radak, Z.; Takeda, M. Lactate metabolism and satellite cell fate. Front. Physiol. 2020, 11, 610983. [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191-1212. [CrossRef]
- Bergman, B.C.; Horning, M.A.; Casazza, G.A.; Wolfel, E.E.; Butterfield, G.E.; Brooks, G.A. Endurance training increases gluconeogenesis during rest and exercise in men. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E244–E251. [CrossRef]
- Bergman, B.C.; Tsvetkova, T.; Lowes, B.; Wolfel, E.E. Myocardial glucose and lactate metabolism during rest and atrial pacing in humans. J. Physiol. 2009, 587, 2087–2099. [CrossRef]
- Glenn, T.C.; Martin, N.A.; Horning, M.A.; McArthur, D.L.; Hovda, D.A.; Vespa, P.; Brooks, G.A. Lactate: Brain fuel in human traumatic brain injury: A comparison with normal healthy control subjects. J. Neurotrauma 2015, 32, 820–832. [CrossRef]
- Suhara, T.; Hishiki, T.; Kasahara, M.; Hayakawa, N.; Oyaizu, T.; Nakanishi, T.; Kubo, A.; Morisaki, H.; Kaelin, W.G. Jr.; Suematsu, M.; Minamishima, Y.A. Inhibition of the oxygen sensor PHD2 in the liver improves survival in lactic acidosis by activating the Cori cycle. Proc. Natl. Acad. Sci. USA 2015, 112, 11642-11647. [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Sign. Transd. Targ. Ther. 2022, 7, 305. [CrossRef]
- Bartolini, B.; Mannelli, M.; Gamberi, T.; Fiaschi, T. The multiple roles of lactate in the skeletal muscle. Cells 2024, 13, 1177. [CrossRef]
- Colegio, O.R.; Shu, N.Q.; Shabo, A.I.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.E.; Philipps, G.M.; Cline, G.W.; Philipps, A.J.; Medzhitov, R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559-563. [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453-463. [CrossRef]
- Ragni, M.; Fornelli, C.; Nisoli, E.; Penna, F. Amino acids in cancer and cachexia: An integrated view. Cancers 2022, 14, 5691. [CrossRef]
- Stalnecker, C.A.; Cluntun, A.A.; Cerione, R.A. Balancing redox stress: Anchorage-independent growth requires reductive carboxylation. Transl. Cancer Res. 2016, 5, S433-S437. [CrossRef]
- Yeo, E.-J. Hypoxia and aging. Exp. Mol. Med. 2019, 51, 67. [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573-587. [CrossRef]
- Wei, Y.; Giunta, S.; Xia, S. Hypoxia in aging and aging-related diseases: Mechanism and therapeutic strategies. Int. J. Mol. Sci. 2022, 23, 8165. [CrossRef]
- Kozlov, A.M.; Lone, A.; Betts, D.H.; Cumming, R.C. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci. Rep. 2020, 10, 8388. [CrossRef]
- Nishimura, K.; Fukuda, A.; Hisatake, K. Mechanisms of the metabolic shift during somatic cell reprogramming. Int. J. Mol. Sci. 2019, 20, 2254. [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [CrossRef]
- Davis, L.; Recktenwald, M.; Hutt, E.; Fuller, S.; Briggs, M.; Goel, A.; Daringer, N. Targeting HIF-2α in the tumor microenvironment: Redefining the role of HIF-2α for solid cancer therapy. Cancers 2022, 14, 1259. [CrossRef]
- Löfstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Overnberger, M.; Pollinger, L.; Påhlman, S. Hypoxia inducible factor-2α in cancer. Cell Cycle 2007, 6, 919–926. [CrossRef]
- de Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [CrossRef]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; Panis, C.; Silva, R.C.M.C.; Abdelhay, E. The hypoxia-inducible factor-1 alpha signaling pathway and its relation to cancer and immunology. Am. J. Immunol. 2014, 10, 215-224. [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Günzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conawey, R.C.; Conawey, J.W.; Kaelin W.G. Jr. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Nat. Acad. Sci. USA 2002, 99, 13459-13464. [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [CrossRef]
- Cyran, A.M.; Zhitkovich, A. HIF1, HSF1, and NRF2: Oxidant-responsive trio raising cellular defenses and engaging immune system. Chem. Res. Toxicol. 2022, 35, 1690–1700. [CrossRef]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [CrossRef]
- Song, M.-Y.; Lee, D.-Y.; Chun, K.-S.; Kim, E.-H. The role of Nrf2/keap1 signaling pathway in cancer metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205-17208. [CrossRef]
- Mustacich, D.; Powis, G. Thioredoxin reductase. Biochem. J. 2000, 346, 1-8.
- Holmgren, A.; Johansson, C.; Berndt, C.; Lönn, M.E.; Hudemann, C.; Lillig, C.H. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem. Soc. Trans. 2005, 33, 1375-1377. [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421-436. [CrossRef]
- Chen, L.; Xing, X.; Zhang, P.; Chen, L.; Pei, H. Homeostatic regulation of NAD(H) and NADP(H) in cells. Genes Dis. 2024, 11, 101146. [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amigo, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; Schoonjans, K.; Menzies, K.J.; Auwerx, J. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; Schoonjans, K.; Schreiber, V.; Sauve, A.A.; Menissier-de Murcia, J.; Auwerx, J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [CrossRef]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiraud, L.; Moullan, N.; Hagberg, C.; Li, W.; Timmers, S.; Imhof, R.; Verbeek, J.; Pujol, A.; van Loon, B.; Viscomi, C.; Zeviani, M.; Schrauwen, P.; Sauve, A.A.; Auwerx, J. Pharmacological Inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014, 19, 1034–1041. [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; Gademann, K.; Rinsch, C.; Schoonjans, K.; Sauve, A.A.; Auwerx, J. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [CrossRef]
- Arnhold, J. Oxidation and reduction of biological material. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 55–97. [CrossRef]
- Diaz-Del Cerro, E.; de Toda, I.M.; Félix, J.; Baca, A.; De la Fuente, M. Components of the glutathione cycle as markers of biological age: An approach to clinical application in aging. Antioxidants 2023, 12, 1529. [CrossRef]
- Lang, C.A.; Naryshkin, S.; Schneider, D.L.; Mills, B.J.; Lindemann, R.D. Low blood glutathione levels in healthy aging adults. J. Lab. Clin. Med. 1992, 120, 720-725.
- Loguercio, C.; Taranto, D.; Vitale, L.M.; Beneduce, F.; Del Vecchio Blanco, C. Effect of liver cirrhosis and age on the glutathione concentration in the plasma, erythrocytes, and gastric mucosa of man. Free Radic. Biol. Med. 1996, 20, 483-488. [CrossRef]
- de Toda, M.; Vida, C.; Garrido, A.; De la Fuente, M. Redox parameters as markers of the rate of aging and predictors of life span. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2020, 75, 613-620. [CrossRef]
- Espinoza, S.E.; Guo, H.; Fedarko, N.; DeZern, A.; Fried, L.P.; Xue, Q.-L.; Leng, S.; Beamer, B.; Walston, J.D. Glutathione peroxidase enzyme activity in aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 505-509. [CrossRef]
- Belrose, J.C.; Xie, Y.F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 2012, 5, 11. [CrossRef]
- Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in brain disorders and aging. Molecules 2022, 27, 324. [CrossRef]
- Yang, B.; Lin, Y.; Huang, Y.; Shen, Y.-Q.; Chen, Q. Thioredoxin (Trx): A redox target and modulator of cellular senescence and aging-related diseases. Redox Biol. 2024, 70, 103032. [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165-1207. [CrossRef]
- Pan, M.; Zhang, F.; Qu, K.; Liu, C.; Zhang, J. TXNIP: A double-edged sword in disease and therapeutic outlook. Oxid. Med. Cell. Longevity 2022, 7805115. [CrossRef]
- Hwang, J.; Suh, H.-W.; Jeon, Y.H.; Hwang, E.; Nguyen, L.T.; Yeom, J.; Lee, S.-G.; Lee, C.; Kim, K.J.; Kang, B.S.; Jeong, J.-O.; Oh, T.-K.; Choi, I.; Lee, J.-O.; Kim, M.H. The structural basis for the negative regulation of thioredoxin by thioredoxin-interacting protein. Nat. Commun. 2014, 5, 2958. [CrossRef]
- Ismael, S.; Nasochi, S.; Li, L.; Aslam, K.S.; Khan, M.M.; El-Remessy, A.B.; McDonald, M.P.; Liao, F.-F.; Ishrat, T. Thioredoxin interacting protein regulates age-associated neuroinflammation. Neurobiol. Dis. 2021, 156, 105399. [CrossRef]
- Choi, E.-H.; Park, S.-J. TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp. Mol. Med. 2023, 55, 1348-1356. [CrossRef]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103. [CrossRef]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; Kahn, B.B.; Cantley, L.C. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; McGraw, T.E.; Wu, N. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell. Rep. 2017, 19, 2005–2013. [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta. 1994, 1219, 26–32. [CrossRef]
- Oberacker, T.; Bajorat, J.; Ziola, S.; Schroeder, A.; Röth, D.; Kastl, L.; Edgar, B.A.; Wagner, W.; Gülow, K.; Krammer, P.H. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. FEBS Lett. 2018, 592, 2297-2307. [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [CrossRef]
- Chen, J.; Saxena, G; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008, 57, 938–944. [CrossRef]
- Gokulakrishnan, K.; Mohanavalli, K.T.; Monickaraj, F.; Mohan, V.; Balasubramanyam, M. Subclinical inflammation/oxidation as revealed by altered gene expression profiles in subjects with impaired glucose tolerance and Type 2 diabetes patients. Mol. Cell. Biochem. 2009, 324, 173–181. [CrossRef]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.-F.; McDonald, M.P.; Ishrat, T. Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer’s disease Brain. J. Alzheimers. Dis. 2019, 68, 255–265. [CrossRef]
- Su, C.-J.; Feng, Y.; Liu, T.-T.; Liu, X.; Bao, J.-J.; Shi, A.-M.; Hu, D.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-interacting protein induced alpha-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [CrossRef]
- Ismael, S.; Wajidunnisa; Sakata, K.; McDonald, M.P.; Liao, F.-F.; Ishrat, T. ER stress associated TXNIP-NLRP3 inflammasome activation in hippocampus of human Alzheimer’s disease. Neurochem. Int. 2021, 148, 105104. [CrossRef]
- Tsubaki, H.; Mendsaikhan, A.; Buyandelger, U.; Tooyama, I.; Walker, D.G. Localization of thioredoxin-interacting protein in aging and Alzheimer’s disease brains. NeuroSci. 2022, 3, 166-185. [CrossRef]
- Kim, S.Y.; Suh, H.-W.; Chung, J.W.; Yoon, S.-R.; Choi, I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell. Mol. Immunol. 2007, 4, 345-351.
- Yu, F.-Y.; Chai, T.F.; He, H.; Hagen, T.; Luo, Y. Thioredoxin-interacting protein (Txnip) gene expression. Sensing oxidative phosphorylation status and glycolytic rate. J. Biol. Chem. 2010, 285, 25822-25830. [CrossRef]
- Fabrizio, P.; Liou, L.-L.; Moy, V.N.; Diaspro, A.; Valentine, J.S.; Gralla, R.B.; Longo, V.D. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 2013, 163, 35-46. [CrossRef]
- Harris, N.; Costa, V.; Maclean, M.; Mollapour, M.; Moradas-Ferreira, P.; Piper, P.W. MnSOD overexpression extends the yeast chronological (G(0)) life span but acts independently of Sir2p histone deacetylase to shorten the replicative life span of dividing cells. Free Radic. Biol. Med. 2003, 34, 1599-1606. [CrossRef]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785-5790. [CrossRef]
- Mao, C.; Yuan, J.-Q.; Lv, Y.-B.; Gao, X.; Yin, Z.-X.; Kraus, V.B.; Luo, J.-S.; Chei, C.-L.; Matchar, D.B.; Zeng, Y.; Shi, X.-M. BMC Genetics 2019, 19, 104. [CrossRef]
- Huie, R.E.; Padmaja, S. Reactions of NO and O2•−. Free Radic. Res. Commun. 1993, 18, 195–199. [CrossRef]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1997, 10, 1285–1292. [CrossRef]
- Nandi, A.; Yan, L.-L. Jana, C.K.; Das, N. Role of catalase in oxidative stress- and age-associated degenerative diseases. Oxid. Med. Cell. Longev. 2019, 2019, 9613090. [CrossRef]
- Wu, M.; Deng, C.; Lo, T.-H.; Chan, K.-Y.; Li, X.; Wong, C.-M. Peroxiredoxin, senescence, and cancer. Cells 2022, 11, 1772. [CrossRef]
- Wonsey, D.R.; Zeller, K.I.; Dang, C.V. The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Prod. Natl. Acad. Sci. USA 2002, 99, 6649-6655. [CrossRef]
- Huh, J.Y.; Kim, Y.; Jeong, J.; Park, J.; Kim, I.; Huh, K.H.; Kim, Y.S.; Woo, H.A.; Rhee, S.G., Lee, K.-J.; Ha, H. Peroxiredoxin 3 is a key molecule regulating adipocyte oxidative stress, mitochondrial biogenesis, and adipokine expression. Antioxid. Redox Signal. 2012, 16, 229-243. [CrossRef]
- Yamamoto-Imoto, H.; Minami, S.; Shioda, T.; Yamashita, Y.; Sakai, S.; Maeda, S.; Yamamoto, T.; Oki, S.; Takashima, M.; Yamamura, T.; Yanagawa, K.; Edahiro, R.; Iwatani, M.; So, M.; Tokumura, A.; Abe, T.; Imamura, R.; Nonomura, N.; Okada, Y.; Ayer, D.E.; Ogawa, H.; Hara, E.; Takabatake, Y.; Isaka, Y.; Nakamura, S.; Yoshimori, T. Age-associated decline of mondoA drives cellular senescence through impaired autophagy and mitochondrial homeostasis. Cell Rep. 2022, 38, 110444. [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 and MTH1 cooperate to prevent ROS-mediated inhibition of telomerase. Genes Dev. 2018, 32, 658-669. [CrossRef]
- Ahmed, W.; Lingner, J. PRDX1 counteracts catastrophic telomeric cleavage events that are triggered by DNA repair activities post oxidative damage. Cell Rep. 2020, 33, 108347. [CrossRef]
- Aeby, E.; Ahmed, W,; Redon, S.; Simanis, V.; Lingner, J. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep. 2016, 17, 3107-3114. [CrossRef]
- Chrichton, R.R.; Ward, R.J. Iron homeostasis. Met. Ions Biol. Syst. 1998, 35, 633–665.
- Ponka, P. Cellular iron metabolism. Kidney Int. 1999, 55, S2–S11. [CrossRef]
- Cankurtaran, M.; Yavuz, B.B.; Halil, M.; Ulger, Z.; Haznedaroglu, I.C.; Anogul, S. increased ferritin levels could reflect ongoing aging-associated inflammation and may obscure underlying iron deficiency in the geriatric population. Eur. Geriat. Med. 2012, 3, 277-280. [CrossRef]
- Kirsipuu, T.; Zadorožnaja, A.; Smirnova, J.; Friedemann, M.; Plitz, T.; Tougu, V.; Palumaa, P. Copper(II)-binding equilibria in human blood. Sci. Rep. 2020, 10, 5686. [CrossRef]
- Cabrera, A.; Alonzo, E.; Sauble, E.; Chu, Y.L.; Nguyen, D.; Linder, M.C.; Sato, D.S.; Mason, A.Z. Copper binding components of blood plasma and organs, and their responses to influx of large doses of (65)Cu, in the mouse. Biometals 2008, 21, 525–543. [CrossRef]
- Osaki, S. Kinetic studies of ferrous ion oxidation with crystalline human ferroxidase (ceruloplasmin). J. Biol. Chem. 1996, 241, 5053–5059. [CrossRef]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.-P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578-6586. [CrossRef]
- Kono, S.; Yoshida, K.; Tomosugi, N.; Terada, T.; Hamaya, Y.; Kanaoka, S.; Miyajima, H. Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 2010, 1802, 968–975. [CrossRef]
- Kono, S. Aceruloplasminemia: An update. Int. Rev. Neurobiol. 2013, 110, 125–151. [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [CrossRef]
- Cheli, V.T.; Sekhar, M.; Santiago Conzález, D.A.; Angeliu, C.G.; Denaroso, G.E.; Smith, Z.; Wang, C.; Paez, P.M. The expression of ceruloplasmin in astrocytes is essential for postnatal myelination and myelin maintenance in the adult brain. Glia 2023, 71, 2323-2342. [CrossRef]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539 –2543. [CrossRef]
- Morita, H.; Ikeda, S.; Yamamoto, K.; Morita, S.; Yoshida, K.; Nomoto, S.; Kato, M.; Yanagisawa, N. Hereditary ceruloplasmin deficiency with hemosiderosis: A clinicopathological study of a Japanese family. Ann. Neurol. 1995, 37, 646–656. [CrossRef]
- Yoshida, K.; Furihata, K.; Takeda, S.; Nakamura, A.; Yamamoto, K.; Morita, H.; Hiyamuta, S.; Ikeda, S.; Shimizu, N.; Yanagisawa, N. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat. Genet. 1995, 9, 267–272. [CrossRef]
- Jeong, S.Y.; David, S. Age-related changes in iron homeostasis and cell death in the cerebellum of ceruloplasmin-deficient mice. J. Neurosci. 2006, 26, 9810-9819. [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165-176. [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369-379. [CrossRef]
- Lu, B.; Chen, X.-b.; Hong, Y.-c.; Zhu, H.; He, Q.-j.; Yang, B.; Ying, M.-d.; Cao, J. Identification of PRDX6 as a regulator of ferroptosis. Acta Pharmacol. Sin. 2019, 40, 1334-1342. [CrossRef]
- Torres-Velarde, J.M.; Allen, K.N.; Salvador-Pascual, A.; Leija, R.G.; Luang, D.; Moreno-Santillán, D.D.; Ensminger, D.C.; Vázquez-Medina, J.P. peroxiredoxin 6 suppresses ferroptosis in lung endothelial cells. Free Radic. Biol. Med. 2024, 218, 82-93. [CrossRef]
- Sauret, J.M.; Marinides, G.; Wang, G.K. Rhabdomyolysis. Am. Fam. Phys. 2002, 65, 907-912.
- Rother, R.P.; Bell, L.; Hillman, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. J. Am. Med. Assoc. 2005, 293, 1653-1662. [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffmann, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198-201. [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and hemopexin in heme detoxification and iron recycling. In: Acute phase proteins – regulation and functions of acute phase proteins; Veas, F., Ed.; Intech: Rijeka, Croatia. 2011; pp. 261-288. [CrossRef]
- Bunn, H.F.; Jandl, J.H. Exchange of heme among hemoglobins and hemoglobin and albumin. J. Biol. Chem. 1968, 243, 465–475. [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobson, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [CrossRef]
- Pan, M.; Wang, P.; Zheng, C.; Zhang, H.Y.; Lin, S.; Shao, B.; Zhuge, Q.; Jin, K. Aging systemic milieu impairs outcome after ischemic stroke in rats. Aging Dis. 2017, 8, 519–530. [CrossRef]
- Sarpong-Kumankomah, S.; Knox, K.B.; Kelly, M.E.; Hunter, G.; Popescu, B.; Nichol, H.; Kopciuk, K.; Ntanda, H.; Galler, J. Quantification of human plasma metalloproteins in multiple sclerosis, ischemic stroke and healthy controls reveals an association of haptoglobin-hemoglobin complexes with age. PLoS ONE 2022, 17, e0262160. [CrossRef]
- Schipper, H.M.; Song, W.; Tavitan, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Progr. Neurobiol. 2019, 172, 40-70. [CrossRef]
- Wei, D.; Qu, C.; Zhao, N.; Li, S.; Pu, N.; Song, Z.; Tao, Y. The significance of precisely regulating heme oxygenase-1 expression: Another avenue for treating age-related ocular disease? Ageing Res. Rev. 2024, 97, 102308. [CrossRef]
- Picard, E.; Ranchon-Cole, I.; Jonet, L.; Beaumont, C.; Behar-Cohen, F.; Courtois, Y.; Jeanny, J.-C. Light-induced retinal degeneration correlates with changes in iron metabolism gene expression, ferritin level, and aging. Invest. Ophthalmol. Vis. Sci. 2011, 52, 1261–1274. [CrossRef]
- Chen, C.; Yang, K.; He, D.; Yang, B.; Tao, L.; Chen, J.; Wu, Y. Induction of ferroptosis by HO-1 contributes to retinal degeneration in mice with defective clearance of all-trans-retinal. Free Radic. Biol. Med. 2023, 194, 245–254. [CrossRef]
- Miyamura, N.; Ogawa, T.; Boylan, S.; Morse, L.S.; Handa, J.T.; Hjelmeland, L.M. Topographic and age-dependent expression of heme oxygenase-1 and catalase in the human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 2004, 45, 1562–1565. [CrossRef]
- Ma, C.; Pi, C.; Yang, Y.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; He, X. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1. PLoS ONE 2017, 12, e0170930. [CrossRef]
- Wu, Y.; Williams, E.G.; Dubuis, S.; Mottis, A.; Jovaisaite, V.; Houton, S.M.; Argmann, C.A.; Faridi, P.; Wolski, W.; Kutalik, Z.; Zamboni, N.; Auwerx, J.; Aebersold, R. Multilayered genetic and omics dissection of mitochondrial activity in a mouse reference population. Cell 2014, 158, 1415–1430. [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; Charpagne, A.; Chong, Y.S.; Descombes, P.; Feng, L.; Francis-Emmanuel, P.; Garratt, E.S.; Giner, M.P.; Green, C.O.; Karaz, S.; Kothandaraman, N.; Marquis, J.; Metairon, S.; Moco, S.; nelson, G.; Ngo, S.; Pleasants, T.; Raymond, F.; sayer, A.A.; Sim, C.M.; Slater-Jefferies, J.; Syddall, H.E.; Tan, P.F.; Titcombe, P.; Vaz, C.; Wetbury, L.D.; Wong, G.; Yonghui, W.; Cooper, C.; Sheppard, A.; Godfey, K.M.; Lillycrop, K.A.; Karnani, N.; Feige, J.N. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; Counts, S.E.; Auwerx, J. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [CrossRef]
- Beck, J.S.; Mufson, E.J.; Counts, S.E. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 610–614. [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [CrossRef]
- Udagawa, O.; Ishihara, T.; Maeda, M.; Matsunaga, Y.; Tsukamoto, S.; Kawano, N.; Miyado, K.; Shitara, H.; Yokota, S.; Nomura, M.; Mihara, K.; Mizushima, N.; Ishihara, N. Mitochondrial fission factor Drp1 maintains oocyte quality via dynamic rearrangement of multiple organelles. Curr. Biol. 2014, 24, 2451–2458. [CrossRef]
- Kageyama, Y.; Zhang, Z.; Roda, R.; Fukaya, M.; Wakabayashi, J.; Wakabayashi, N.; Kensler, T.W.; Reddy, H.; Iijima, M.; Sesaki, H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012, 197, 535–551. [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373-429. [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structure, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695 55-72. [CrossRef]
- López-Otin, C.; Blasco, M. A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell, 2013, 153, 1194-1217. [CrossRef]
- Calderwood, S.K.; Murshid, A.; Prince, T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology, 2009, 55, 550-558. [CrossRef]
- Löw, P. The role of ubiquitin-proteasome system in aging. Gen. Comp. Endocrinol. 2011, 172, 39-43. [CrossRef]
- Saez, I.; Vilchez, D. The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics 2014, 15, 38-51. [CrossRef]
- Frankowska, N.; Lisowska, K.; Witkowski, J.M. Proteolysis dysfunction in the process of aging and age-related diseases. Front. Aging 2022, 3, 927630. [CrossRef]
- Ponnappan, U.; Zhong, M.; Trebilcock, G.U. Decreased proteasome-mediated degradation in T cells from the elderly: A role in immune senescence. Cell. Immunol. 1999, 192, 167–174. [CrossRef]
- Chondrogianni, N.; Stratford, F.L.L.; Trougakos, I.P.; Friguet, B.; Rivett, A.J.; Gonos, E.S. Central role of the proteasome in senescence and survival of human fibroblasts. Induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J. Biol. Chem. 2003, 278, 28026–28037. [CrossRef]
- Dahlmann, B. Role of proteasomes in disease, BMC Biochem. 2007, 8 (suppl. 1) S3. [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728-741. [CrossRef]
- Kobayashi, S. Choose delicately and reuse adequately: The newly revealed process of autophagy. Biol. Pharmaceut. Bull. 2015, 38, 1098-1103. [CrossRef]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.G.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.K.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333-345. [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309-314. [CrossRef]
- García-Prat, L.; Martinez-Vicente, M.; perdiguero, E.; ortet, L.; Rodriguez-Ubreva, J.; Rebello, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; Sandri, M.; Muñoz-Cánoves, P. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662-3670. [CrossRef]
- Pinti, M.; Cevenini, E.; Nasi, M.; de Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; Trenti, T.; Franceschi, C.; Cossarizza, A. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging”. Eur. J. Immunol. 2014, 44, 1552-1562. [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; narenda, D.P.; Borsche, M.; Klein, C.; Youle, R.J. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [CrossRef]
- Penna, F.; Ballarò, R.; Beltrá, M.; de Lucia, S.; Costelli, P. Modulating metabolism to improve cancer-imduced muscle wasting. Oxid. Med. Cell. Longevity 2018, 7153610. [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14,754-762. [CrossRef]
- Bye, A.; Sjøblom, B.; Wentzel-Larsen, T.; Grønberg; B.H.; baracos, V.E.; Hjemstad, M.J.; Aass, N.; Bremnes, R.M.; Fløtten, Ø.; Jordhøy, M. Muscle mass and association to quality of life in non-small cell lung cancer patients. J. Cachexia, Sarcopenia Muscle 2017, 8, 759-767. [CrossRef]
- Nipp, R.D.; Fuchs, G.; El-Jawahri, A.; Mario, J.; Troschel, E.M.; Greer, J.A.; Gallgher, E.R.; Jackson, V.A.; Kambadakone, A.; Hong, T.S.; Temel, J.S.; Fintelmann, F.J. Sarcopenia is associated with quality of life and depression in patients with advanced cancer. Oncologist 2017, 23, 97–104. [CrossRef]
- Morley, J.E.; Anker, S.D.; Evans, W.J. Cachexia and aging: An update based on the fourth international cachexia meeting. J. Nutr. Health Aging 2009, 13, 47-55. [CrossRef]
- Ali, S.; Garcia, J.M. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options. Gerontology 2014, 60, 294-305. [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transd. Targ. Ther. 2023, 8, 239. [CrossRef]
- De la Fuente, M.; Miquel, J. An update of the oxidation-inflammation theory of aging: The involvement of the immune system in oxi-inflamm-aging. Curr. Pharm. Des. 2009, 15, 3003–3026. [CrossRef]
- Martinez de Toda, I.; Ceprian, N.; Diaz-Del Cerro, E.; De la Fuente, M. The role of immune cells in oxi-inflamm-aging. Cells 2021, 10, 2974. [CrossRef]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: Age-related diseases or longevity? Ageing Res. Rev. 2021, 71, 101422. [CrossRef]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [CrossRef]
- Miao, E.A; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirios, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; Espevik, T.; Lien, E.; Fitzgerald, K.A.; Rock, K.L.; Moore, K.J.; Wright, S.D.; Hornung, V.; Latz, E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [CrossRef]
- Ward, R.J; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [CrossRef]
- Pittet, D.; Wenzel, R.P. Nosocomial bloodstream infections. Secular trends in rates, mortality, and contribution to total hospital deaths. Arch. Intern. Med. 1995, 155, 1177–1184. [CrossRef]
- Llewelyn, M.J.; Cohen, J. Tracking the microbes in sepsis: Advancements in treatment bring challenges for microbial epidemiology. Clin. Infect. Dis. 2007, 44, 1343–1348. [CrossRef]
- Kollef, K.E.; Schramm, G.E.; Wills, A.R.; Reichley, R.M.; Mirek, S.T.; Kollef, M.H. Predictors of 30-day mortality and hospital costs in patients with ventilator-associated pneumonia attributed to potentially antibiotic-resistant gram-negative bacteria. Chest 2008, 134, 281–287. [CrossRef]
- Babu, M.; Menon, V.P.; Devi, P.U. Prevalence of antimicrobial resistant pathogens in severe sepsis and septic shock patients. J. Young Pharm. 2018, 10, 358–361. [CrossRef]
- Lin, G.-L.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and immune pathogenesis of viral sepsis. Front. Immunol. 2018, 9, 2147. [CrossRef]
- Podnos, Y.D.; Jiminez, J.C.; Wilson, S.E. Intraabdominal sepsis in elderly persons. Clin. Infect. Dis. 2002, 35, 62–68. [CrossRef]
- Williams, M.D.; Braun, L.A.; Cooper, I.M.; Johnston, J.; Weiss, R.V.; Qualy, R.I.; Linde-Zwirble, W. Hospitalized cancer patients with severe sepsis: Analysis of incidence, mortality, and associated costs of care. Crit. Care 2004, 8, R291–R298. [CrossRef]
- Steinberg, D.; Parthasarathy, S.; Carew, T.E.; Khoo, J.C.; Witztum. Beyond cholesterol: Modification of low-density lipoprotein that increases its atherogenicity. N. Engl. J. Med. 1989, 320, 915-924. [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233-241. [CrossRef]
- Wick, G.; Knoflach, M, Xu, Q. Autoimmune and inflammatory mechanisms in atherosclerosis. Annu. Rev. Atheroscl. 2004,22, 361-403. [CrossRef]
- Tabas, I.; Garcia-Gardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13-22. [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis. Current view and future perspective on lipoprotein modification treatment. Neth. Heart 2017, 25, 231-242. [CrossRef]
- Gisterå, A.; Hansson, G. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368-380. [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis. Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245-259. [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The aging risk and atherosclerosis: A fresh look at arterial homeostasis. Front. Genet. 2017, 8, 216. [CrossRef]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The molecular biomarkers of vascular aging and atherosclerosis: Telomere length and mitochondrial DNA4977 common deletion. Mut. Res. 2020, 784, 108309. [CrossRef]
- Wong, J.J.; Hong, R.; Teo, L.L.Y.; Tan, R.-S.; Koh, A.S. Atherosclerotic cardiovascular disease in aging and the role of advanced cardiovascular imaging. Cardiovasc. Health 2024, 1, 11. [CrossRef]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437-444. [CrossRef]
- Hazen, S.L.; Heinecke, J.W. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J. Clin. Investig. 1997, 99, 2075-2081. [CrossRef]
- Malle, E.; Marsche, G.; Arnhold, J.; Davies, M.J. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim. Biophys. Acta 2006, 1761, 392-415. [CrossRef]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercelotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscl. Thromb. 1991, 11, 1700-1711. [CrossRef]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercelotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879-887. [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon III, R.O.; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383-1389. [CrossRef]
- Eich, R.F.; Li, T.; Lemon, D.D.; Doherty, D.H.; Curry, S.R.; Aitken, J.F.; Mathews, A.J.; Johnson, K.A.; Smith, R.D.; Philips, Jr. G.N.; Olson, J.S. Mechanisms of NO-induced oxidation of myoglobin and hemoglobin. Biochemistry 1996, 35, 6076-6983. [CrossRef]
- Olson, J.S.; Foley, E.W.; Rogge, C.; Tsai, A.L.; Doyle, M.P.; Lemon, D.D. NO scavenging and the hypertensive effect of hemoglobin-based blood substitutes. Free Radic. Biol. Med. 2004, 36, 685-697. [CrossRef]
- Flögel, U.; Fago, A.; Rassaf, T.; Keeping the heart in balance: The functional interactions of myoglobin with nitrogen oxides. J. Exp. Biol. 2010, 213, 2726-2733. [CrossRef]
- Eiserich, J.P.; Baldus, S.; Brennan, M.-L.; Ma, W.; Zhang, C.; Tousson, A.; Castro, L.; Lusis, A.J.; Nauseef, W.M.; White, C.R.; Freeman, B.A. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 2002, 196, 2391–2394. [CrossRef]
- Ariesen, M.J.; Claus, S.P.; Rinkel, G.J.; Algra, A. Risk factors for intracerebral hemorrhage in the general population: A systematic view. Stroke 2003, 34, 2060-2065. [CrossRef]
- Jani, B.; Rajkumar, C. Ageing and vascular ageing. Postgrad. Med. J. 2006, 82, 357-362. [CrossRef]
- Dastur, C.K.; Yu, W. Current management of spontaneous intracerebral haemorrhage. Stroke Vasc. Neurol. 2017, e000047. [CrossRef]
- Flemmig, J.; Schlorke, D.; Kühne, F.-W.; Arnhold, J. Inhibition of heme-induced hemolysis of red blood cells by the chlorite-based drug WF10. Free Rad. Res. 2016, 50, 1386-1395. [CrossRef]
- Deuel, J.W.; Schaer, C.A.; Boretti, F.S.; Opitz, L.; Garcia-Rubio, I.; Baek, J.H.; Spahn, D.R.; Buehler, P.W.; Schaer, D.J. Hemoglobinuria-related acute kidney injury is driven by intrarenal oxidative reactions triggering a heme toxicity response. Cell Death Dis. 2016, 7, e2064. [CrossRef]
- Rosendaal, F.R.; van Hylckama Vlieg A.; Doggen, C.J.M. Venous thrombosis in the elderly. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 310-317. [CrossRef]
- Engbers, M.J.; van Hylckama Vlieg A.; Rosendaal, F.R. Venous thrombosis in the elderly: Incidence, risk factors and risk groups. J. Thromb. Haemost. 2010, 8, 2105-2112. [CrossRef]
- Akrivou, D.; Perlepe, G.; Kirgou, P.; Gourgoulianis, K.I.; Malli, F. Pathophysiological aspects of aging in venous thromboembolism: An update. Medicina 2022, 58, 1078. [CrossRef]
- Sinclair, A.; Dunning, T.; Rodriguez-Mañas, L. Diabetes in older people: New insights and remaining challenges. Lancet Diabet. Endocrinol. 2015, 3, 275-285. [CrossRef]
- Arnhold, J. Cell and tissue destruction in selected disorders. In Cell and Tissue Destruction. Mechanisms, Protection, Disorders; Academic Press: London, UK; San Diego, CA, USA; Cambridge, MA, USA; Oxford, UK, 2020; pp. 249–287. [CrossRef]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94-112. [CrossRef]
- Hirose, A.; Tanikawa, T.; Mori, H.; Okada, Y.; Tanaka, Y. Advanced glycation end products increase endothelial permeability through RAGE/Rho signaling pathway. FEBS Lett. 2010, 584, 61-66. [CrossRef]
- Koenig, R.J.; Peterson, C.M.; Jones, R.L.; Saudek, C.; Lehrmann, M.; Cerami, A. Correlation of glucose regulation and hemoglobin A1c in diabetes mellitus. N. Engl. J. Med. 1976, 295, 417-420. [CrossRef]
- Bunn, H.F.; Gabbay, K.H.; Gallop, P.M. The glycosylation of hemoglobin: Relevance to diabetes mellitus. Science 1978, 200, 21-27. [CrossRef]
- Shin, S.; Ku, Y.-H.; Ho, J.-X.; Kim, Y.-K.; Suh, J.-S.; Singh, M. Progressive impairment of erythrocyte deformability as indicator of microangiopathy in type 2 diabetes mellitus. Clin. Hemorheol. Microcirc. 2007, 36, 253-261.
- Babu, N.; Singh, M. Influence of hyperglycemia on aggregation, deformability and shape parameters of erythrocytes. Clin. Hemorheol. Microcirc. 2004, 31, 273-280.
- Kung, C.-M.; Tseng, Z.-I.; Wang, H.-L. Erythrocyte fragility with level of glycosylated hemoglobin in type 2 diabetic patients. Clin. Hemorheol. Microcirc. 2009, 43, 345-351. [CrossRef]
- Agarwal, R.; Smart, T.; Nobre-Cordosa, J.; Richards, C.; Bhatnagar, R.; Tufail, A. Shima, D.; Jones, P.H.; Pavesio, C. Assessment of red blood cell deformability in type 2 diabetes mellitus and diabetic retinopathy by dual optical tweezers stretching technique. Sci. Rep. 2016, 6, 15873. [CrossRef]
- Ernst, E.; Matrai, A. Altered red and white blood cell rheology in type II diabetes. Diabetes 1986, 35, 1412-1415. [CrossRef]
- Popov, D. Endothelial cell dysfunction in hyperglycemia: Phenotypic change, intracellular signaling modification, ultrastructural alteration, and potential clinical outcomes. Int. J. Diabetes Mellitus 2010, 2, 189-195. [CrossRef]
- Popov, D. Towards understanding the mechanisms of impeded vascular function in the diabetic kidney. In Simionescu, M.; Sima, A.; Popov, D. (Eds.) Cellular dysfunction in atherosclerosis and diabetes – Reports from bench to bedside. Publishing House Rom, Acad. Bucharest, 2004, pp. 244-259.
- Dokken, B.B. The pathophysiology of cardiovascular disease and diabetes: Beyond blood pressure and lipids. Diabetes Spectr. 2008, 21, 160-165. [CrossRef]
- Xiang, Y.; Cheng, J.; Wang, D.; Hu, X.; Xie, Y.; Stitham, J.; Atteva, G.; Du, J.; Tang, W.H.; Lee, S.H.; Leslie, K.; Spollett, G.; Liu, Z.; Herzog, E.; Herzog, R.I.; Lu, J.; Martin, K.A.; Hwa, J. Hyperglycemia repression of miR-24 coordinately upregulates endothelial cell expression and secretion of von Willebrand factor. Blood 2015, 125, 3377-3387. [CrossRef]
- Whincup, P.H.; Danesh, J.; Walker, M.; Lennon, L.; Thomson, A.; Appleby, P.; Rumley, A.; Lowe, G.D. Von Willebrand factor and coronary heart disease: Prospective study and meta-analysis. Eur. Heart J. 2002, 23, 1764-1770. [CrossRef]
- Frankel, D.S.; Meigs, J.B.; Massaro, J.M.; Wilson, P.W.; O’Donnell, C.J.; D’Agostino, R.B.; Tofler, G.H.; Von Willebrand factor, type 2 diabetes mellitus, and risk of cardiovascular disease: The Framingham offspring study. Circulation 2008, 118, 2533-2539. [CrossRef]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332. [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [CrossRef]
- Nakamura, K.; Smyth, M.J. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol. Immunol. 2020, 17, 1–12. [CrossRef]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune response. Front. Immunol. 2020, 11, 940. [CrossRef]
- Barnestein, R.; Galland, L.; Kalfeist, L.; Ghiringhelli, F.; Ladoire, S.; Limagne, E. Immunosuppressive tumor microenvironment modulation by chemotherapies and targeted therapies to enhance immunotherapy effectiveness. OncoImmunology 2022, 11, 2120676. [CrossRef]
- Tie, Y.; Tang, F.; Wei, Y.-Q.; Wie, X.-W. Immunosuppressive cells in cancer: Mechanisms and potential therapeutic targets. J. Hematol. Oncol. 2022, 15, 61. [CrossRef]
- Othman, N.; Jamal, R.; Abu, N. Cancer-derived exosomes as effectors of key inflammation-related players. Front. Immunol. 2019, 10, 2103. [CrossRef]
- He, Z.; Zhang, S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front. Immunol. 2021, 12, 741305. [CrossRef]
- Pastorevka, S.; Ratcliffe, P.J.; Pastorek, J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. J. Compil. 2008, 101 (Suppl. 4) 8-15. [CrossRef]
- Kaluz, S.; Kaluzová, M.; Liao, S.-Y.; Lerman, M.; Stanbridge, E.J. Transcriptional control of the tumor- and hypoxia-marker carbonic anhydrase 9: A one transcription factor (HIF-1) show? Biochim. Biophys. Acta 2009, 1795, 162-177. [CrossRef]
- Becker, H.M. Carbonic anhydrase IX and acid transport in cancer. Br. J. Cancer 2020, 122, 157-167. [CrossRef]
- Zheng, T.; Jäättelä, Liu, B. pH gradient reversal fuels cancer progression. Int. J. Biochem. Cell Biol. 2020, 125, 105796. [CrossRef]
- Park, Y.; Jeong, J.; Seong, S.; Kim, W. In silico evaluation of natural compounds for an acidic extracellular environment in human breast cancer. Cells 2021, 10, 2673. [CrossRef]
- Logozzi, M.; Spugnini, E.; Mizzoni, D.; Di Raimo, R.; Fais, S. Extracellular acidity and increased exosome release as key phenotypes of malignant tumors. Cancer Metastasis Rev. 2019, 38, 93–101. [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227-235. [CrossRef]
- Walti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Bockmann, A.; Guntert, P.; Meier, B.H.; Riek, R. Atomic-resolution structure of a disease-relevant Abeta(1-42) amyloid fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976-E4984. [CrossRef]
- Fezoui, Y.; Teplow, D.B. Kinetic studies of amyloid-bate protein fibril assembly. Differential effects of alpha-helix stabilization. J. Biol. Chem. 2002, 277, 36948-36954. [CrossRef]
- Uversky, V.N. Protein misfolding in lipid-mimetic environments. Adv. Exp. Med. Biol. 2015, 855, 33-66. [CrossRef]
- Prasad, H.; Rao, R. Endosomal acid-base homeostasis in neurodegenerative diseases. Rev. Physiol. Biochem. Pharmacol. 2020, 185, 195-231. [CrossRef]
- Decker, Y.; Németh, E.; Schomburg, R.; Chemla, A.; Fülöp, L.; Menger, M.D.; Liu, Y.; Fassbender, K. Decreased pH in the aging brain and Alzheimer’s disease. Neurobiol. Aging 2021, 101, 40-49. [CrossRef]
- Hagihara, H.; Miyakawa, T. Decreased brain pH correlated with progression of Alzheimer disease neuropathology: A systematic review and meta-analyses of postmortem studies. Int. J. Neuropsychopharmacol. 2024, pyae047. [CrossRef]
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [CrossRef]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial iron homeostasis and its dysfunction in neurodegenerative disorders. Mitochondrion 2015, 21, 92-105. [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.; Noel, K.; Jiang, X.; Linkermann, A.; Murphy, M.E.; Overholtzer, M.; Oyagi, A.; Pagnussat, G.; Park, J.; Ran, Q.; Rosenfeld, C.S.; Salnikow, K.; Tang, D.; Torti, F.; Torti, S.; Toyokuni, S.; Woerpel, K.A.; Zhang, D.D. Ferroptosis: A regulated death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273-285. [CrossRef]
- De Bie, P.; Muller, P.; Wijmenga, C.; Klomp, L.W.J. Molecular pathogenesis of Wilson and Menkes disease: Correlation of mutations with molecular defects and disease phenotypes. J. Med. Genet. 2007, 44, 673–688. [CrossRef]
- Squitti, R.; Pasqualetti, P.; Dal Forno, G.; Moffa, F.; Cassetta, E.; Lupoi, D.; Vernieri, F.; Rossi, L.; Baldassini, M.; Rossini, P.M. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology 2005, 64, 1040–1046. [CrossRef]
- Squitti, R.; Bressi, F.; Pasqualetti, P.; Bonomini, C.; Ghidoni, R.; Binetti, G.; Cassetta, E.; Moffa, F.; Ventriglia, M.; Vernieri, F.; Rossini, P.M. Longitudinal prognostic value of serum “free” copper in patients with Alzheimer disease. Neurology 2009, 72, 50–55. [CrossRef]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, cellular and clinical aspects of intracerebral hemorrhage: Are the enemies within? Curr. Neuropharmacol. 2016, 14, 392–402. [CrossRef]
- Chiabrando, D.; Fiorito, V.; Petrillo, S.; Tolosano, E. Unraveling the role of heme in neurodegeneration. Front. Neurosci. 2018, 12, 712. [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercelotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 13, 377-390. [CrossRef]
- Chiziane, E.; Telemann, H.; Krueger, M.; Adler, J.; Arnhold, J.; Alia, A.; Flemmig, J. Free heme and amyloid-β: A fatal liaison in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 61, 963-984. [CrossRef]
- Song, I.-U.; Kim, Y.-D.; Chung, S.-W.; Cho, H.J. Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern. Med. 2015, 54, 453-457. [CrossRef]
- Ashraf, A.; Clark, M.; So, P.-W. The aging of iron man. Front. Aging Neurosci. 2018, 10, 65. [CrossRef]
- Moshage, H. Cytokines and the hepatic acute phase response. J. Pathol. 1997, 181, 74-80. [CrossRef]
- Lin, T.; Maita, D.; Thundivalappil, S.R.; Riley, F.E.; Hambsch, J.; van Marter, L.J.; Christou, H.A.; Berra, L.; Fagan, S.; Christiani, D.C.; Warren, H.S. Hemopexin in severe inflammation and infection: Mouse models and human diseases. Crit. Care 2015, 19, 166. [CrossRef]
Figure 1.
Role of molecular patterns, tissue damage, and cytotoxic agents during inflammation. The further fate of inflammation depends on the individual state of protecting mechanisms to antagonize cytotoxic agents. Resolution of inflammation is induced when no further damage occurs. As long as novel DAMPs are released from undergoing immune and tissue cells at inflammatory sites, the inflammation persists (presented in blue). Abbreviations: DAMPs?damage-associated molecular patterns, PAMPs?pathogen-associated molecular patterns.
Figure 1.
Role of molecular patterns, tissue damage, and cytotoxic agents during inflammation. The further fate of inflammation depends on the individual state of protecting mechanisms to antagonize cytotoxic agents. Resolution of inflammation is induced when no further damage occurs. As long as novel DAMPs are released from undergoing immune and tissue cells at inflammatory sites, the inflammation persists (presented in blue). Abbreviations: DAMPs?damage-associated molecular patterns, PAMPs?pathogen-associated molecular patterns.
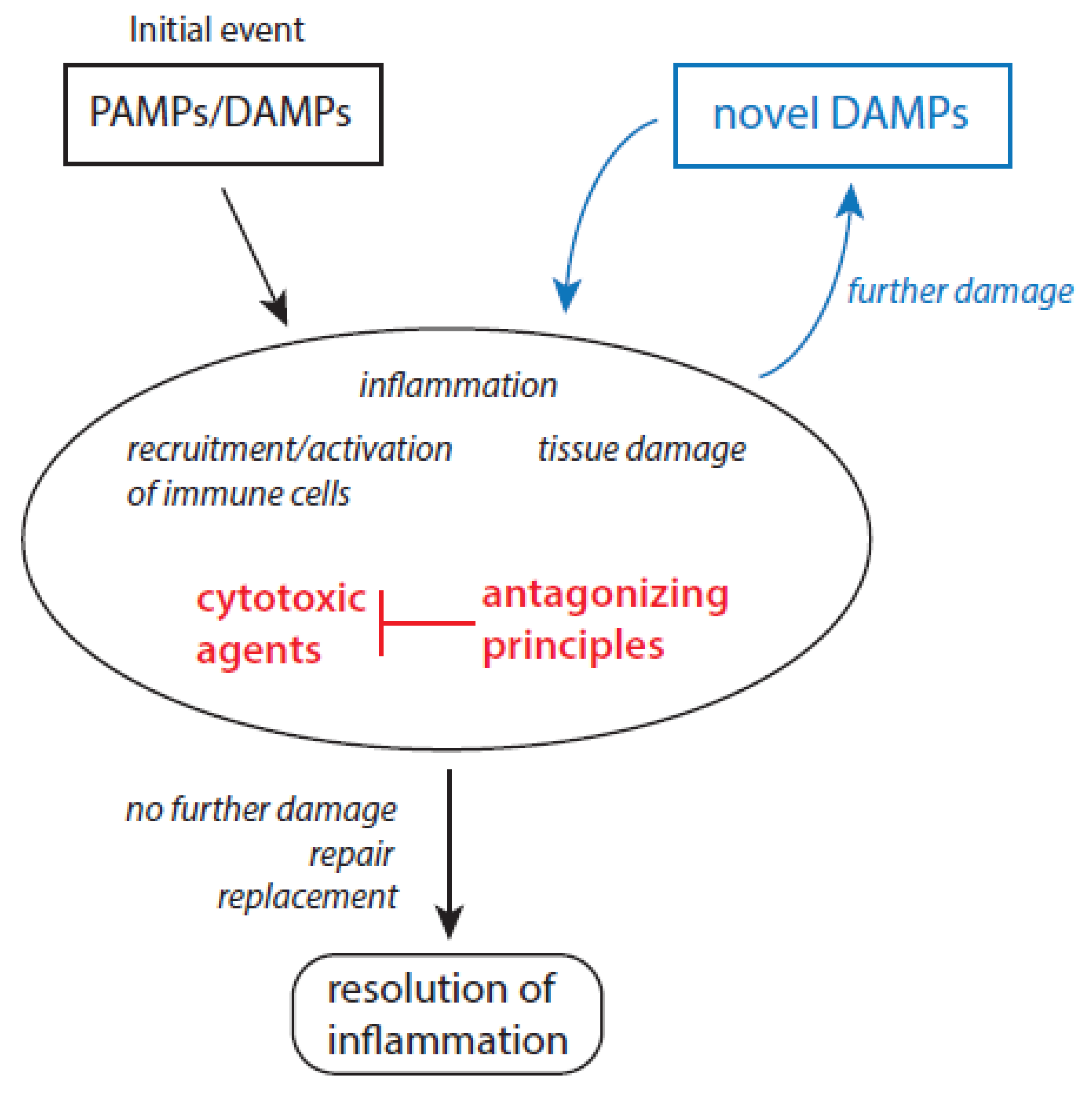
Figure 2.
Possible alterations of the energy metabolism in stressed cells. Potential energy fuels are presented on grey background. Concerning amino acids as energy substrate only glutamate is included. All other amino acids are also converted into intermediate products, which are applied at different places in the TCA cycle. In hypoxic cells, glycolysis is highly up-regulated with lactate as final product (red arrow). Lactate can be used by neighbored cells as fuel to increase mitochondrial processes of energy production (blue arrows). In stressed cells, dysfunctional mitochondria are a source for enhanced production of oxidant-based cytotoxic agents (given in red) starting with superoxide anion radicals, and followed by hydrogen peroxide, ferrous ions, hydroxyl radicals, and peroxynitrite. Further explanations are given in the text.
Figure 2.
Possible alterations of the energy metabolism in stressed cells. Potential energy fuels are presented on grey background. Concerning amino acids as energy substrate only glutamate is included. All other amino acids are also converted into intermediate products, which are applied at different places in the TCA cycle. In hypoxic cells, glycolysis is highly up-regulated with lactate as final product (red arrow). Lactate can be used by neighbored cells as fuel to increase mitochondrial processes of energy production (blue arrows). In stressed cells, dysfunctional mitochondria are a source for enhanced production of oxidant-based cytotoxic agents (given in red) starting with superoxide anion radicals, and followed by hydrogen peroxide, ferrous ions, hydroxyl radicals, and peroxynitrite. Further explanations are given in the text.
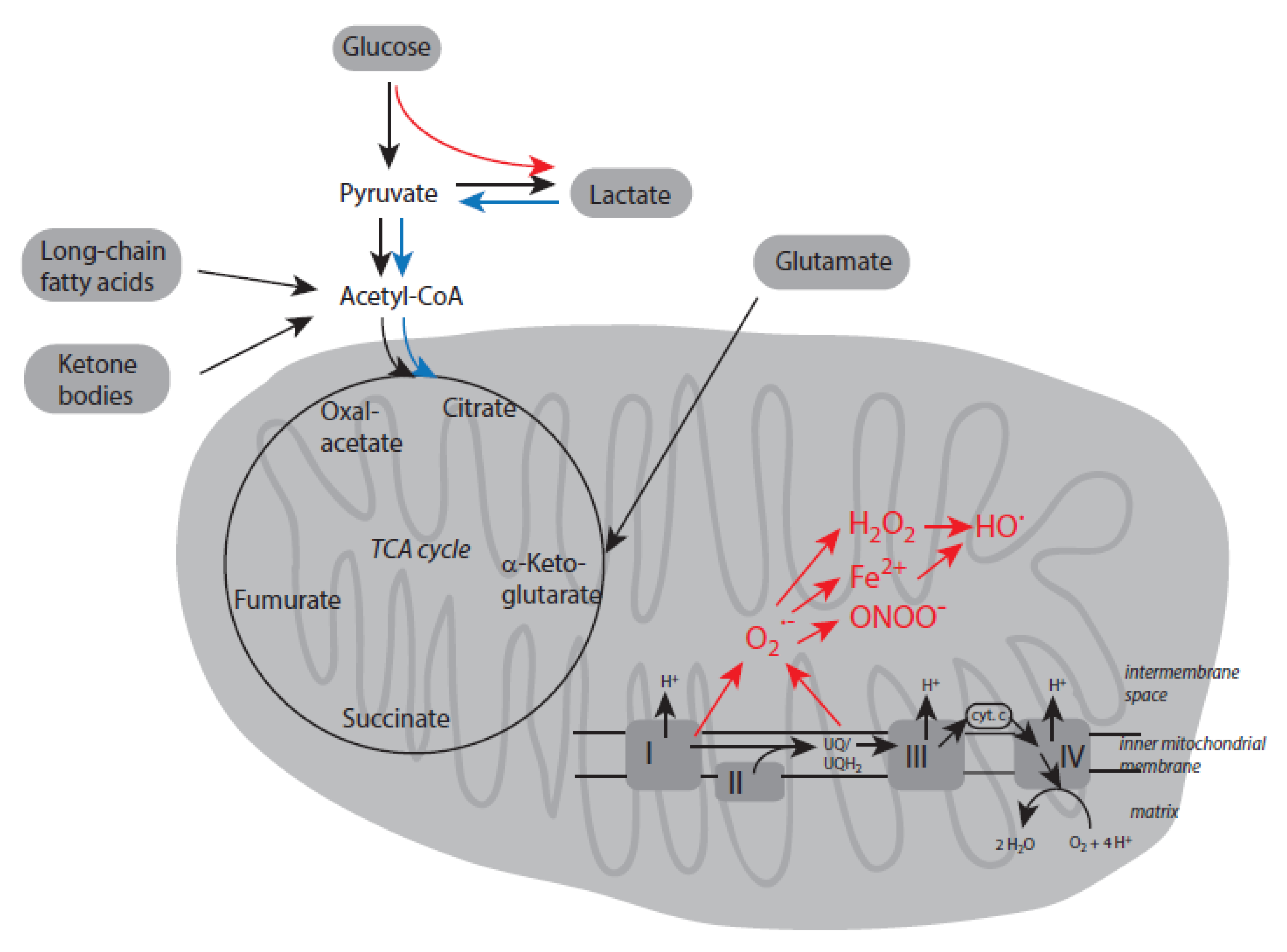
Figure 3.
Basic mechanisms of redox regulation in cells. Metabolites that dominate in resting cells are given in red. The enhanced formation of reactive species like superoxide anion radicals and hydrogen peroxide under oxidative stress as well as the following increase of other redox metabolites are indicated by blue arrows. Abbreviations: ASK-1?apoptosis-signal-regulating kinase 1, GSH?glutathione, GS-SG?oxidized glutathione, LOH?lipid alcohol, LOOH?lipid hydroperoxide, Prx?peroxiredoxin, PTEN?phosphatase and tensin homolog, Trx?thioredoxin, Txnip?thioredoxin interacting protein.
Figure 3.
Basic mechanisms of redox regulation in cells. Metabolites that dominate in resting cells are given in red. The enhanced formation of reactive species like superoxide anion radicals and hydrogen peroxide under oxidative stress as well as the following increase of other redox metabolites are indicated by blue arrows. Abbreviations: ASK-1?apoptosis-signal-regulating kinase 1, GSH?glutathione, GS-SG?oxidized glutathione, LOH?lipid alcohol, LOOH?lipid hydroperoxide, Prx?peroxiredoxin, PTEN?phosphatase and tensin homolog, Trx?thioredoxin, Txnip?thioredoxin interacting protein.
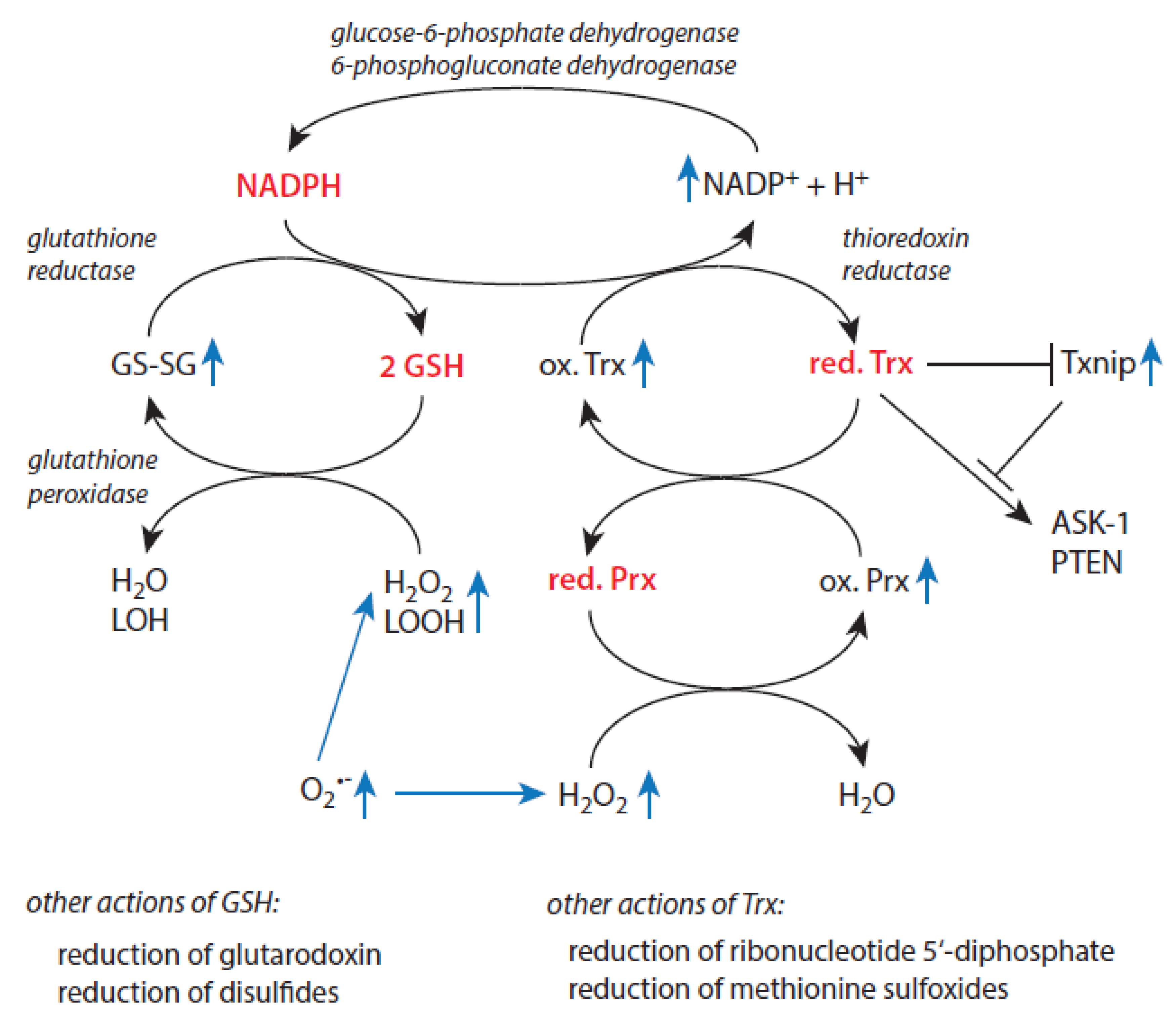

Table 1.
Key features and properties of the two main phases of an acute inflammation.
| Predominant Features or Properties | Initiation and Propagation of Inflammation | Resolution of Inflammation |
|---|---|---|
| Immune response | Recruitment and activation of immune cells | Deactivation of immune cells, immunosuppression |
| Presence of molecular patterns | yes | no |
| Main physiological processes | Elimination of pathogens, removal of damaged cells and cell debris | Repair processes, synthesis of novel extracellular matrix |
| Typical mediators | IL-1, IL-6, IL-15, IL-8, TNF-α | TGF-β, IL-10, VEGF, lipoxins |
| Presence of CRP and SAA | enhanced values | decreasing values |
| Macrophage subtypes | M1 type | M2 type |
| Major pathways for ATP production in macrophages | Glycolysis | oxidative phosphorylation |
| Presence of MDSCs | low | enhanced |
Table 2.
Neutrophil-derived cytotoxic agents and their antagonizing principles.
| Cytotoxic Agent | Antagonizing Principles | References |
|---|---|---|
| Superoxide anion radicals | Superoxide dismutases | [58,59,60,61] |
| Hydrogen peroxide | Catalase, peroxiredoxins, glutathione peroxidases | [62,63,64] |
| Hydroxyl radicals | Limited protection by carbohydrates | [65] |
| Free transition metal ions | Proper control over all aspects of iron and copper ion metabolism | [66,67,68,69,70] |
| Myeloperoxidase | Ceruloplasmin | [71,72,73,74] |
| Hypochlorous acid, hypobromous acid | SCN-, taurine, glutathione, ascorbate | [75,76,77] |
| Hypothiocyanite | glutathione | [78] |
| Elastase | α1-antitrypsin (A1AT), secretory leukocyte protease inhibitor (SLPI), elafin, serpin B1, α2-macroglobulin | [79,80,81,82,83] |
| Cathepsin G | A1AT, SLPI, α1-antichymotrypsin | [79,80,81,84,85,86] |
| Proteinase 3 | A1AT, elafin | [79,80,82,83] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



