
Preprint
Review
The Molecular Intersection of NEK1, C21ORF2, Cyclin F, and VCP in ALS Pathogenesis
This version is not peer-reviewed.


Submitted:
17 March 2025
Posted:
18 March 2025
You are already at the latest version
A peer-reviewed article of this preprint also exists.
Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder characterized by the progressive degeneration of motor neurons, leading to muscle weakness, paralysis, and death. Although significant progress has been made in understanding ALS, its molecular mechanisms remain complex and multifactorial. This review explores the potential convergent mechanisms underlying ALS pathogenesis, focusing on the roles of key proteins including NEK1, C21ORF2, Cyclin F, VCP, and TDP-43. Recent studies suggest that mutations in C21ORF2, lead to the stabilization of NEK1, while Cyclin F mutations activate VCP, resulting in TDP-43 aggregation. TDP-43 aggregation, a hallmark of ALS, impairs RNA processing and protein transport, both of which are essential for neuronal function. Furthermore, TDP-43 has emerged as a key player in DNA damage repair, translocating to DNA damage sites and recruiting repair proteins. Given that NEK1, VCP, and Cyclin F are also involved in DNA repair, this review examines how these proteins may intersect to disrupt DNA damage repair mechanisms, contributing to ALS progression. Impaired DNA repair and protein homeostasis are suggested to be central downstream mechanisms in ALS pathogenesis. Ultimately, understanding the interplay between these pathways could offer novel insights into ALS and provide potential therapeutic targets. This review aims to highlight the emerging connections between protein aggregation, DNA damage repair, and cellular dysfunction in ALS, fostering a deeper understanding of its molecular basis and potential avenues for intervention.
Keywords:
amyotrophic lateral sclerosis (ALS)
; TDP-43 aggregation
; NEK1
; C21ORF2
; Cyclin F
; VCP
; DNA damage repair
; protein homeostasis
; neurodegeneration
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative condition marked by the progressive degeneration of both upper and lower motor neurons, ultimately leading to muscle weakness, paralysis, and mortality, primarily due to respiratory failure [1]. Advances in genetic analysis, applied to both familial ALS (fALS) and sporadic ALS (sALS), have enabled the identification of over 50 causative and risk-associated genes for ALS [2]. Functional studies of these genes, particularly those identified earlier such as SOD1, TARDBP (which encodes TDP-43), FUS, and C9ORF72, combined with patient sample analysis and disease models, have provided essential insights into the molecular mechanisms underpinning ALS, encompassing the dysregulation of DNA, RNA, and proteins [3].
Given that abnormal protein aggregation is a hallmark of neurodegenerative disorders [4,5], research on protein misregulation surpasses that of DNA and RNA. For instance, mutations in genes such as SOD1, TARDBP, FUS, and C9ORF72 have been shown to disrupt protein homeostasis by promoting protein aggregation. Mutant SOD1 forms cytotoxic aggregates that impair mitochondrial function [6,7,8], while hexanucleotide repeat expansions (HRE) in a non-coding region of C9ORF72 generate dipeptide repeat proteins (DPRs) that self-assemble, sequestering RNA-binding proteins and proteins with low-complexity sequence domains (LCDs). This impairs the function of RNA-rich, membrane-less organelles such as nucleoli and stress granules, as well as nucleocytoplasmic transport mediated by proteins with LCDs [9,10,11]. Similarly, mutations in TARDBP and FUS increase the formation of cytoplasmic inclusions of TDP-43 and FUS [12,13,14,15], compromising several cellular activities, including protein nucleocytoplasmic transport [16], chaperoning [17], and degradation [18], by sequestering proteins essential for these critical processes.
The role of RNA misregulation in ALS has been suggested by the fact that several ALS-associated proteins, including TDP-43, FUS, and Matrin-3, are RNA-binding proteins [19]. Studies on these proteins have demonstrated that dysregulation of mRNA splicing, transport, and stability contributes to the degeneration of motor neurons [20]. Additionally, C9ORF72 HREs generate toxic RNA foci that may be implicated in ALS pathogenesis [21,22,23,24].
In contrast to protein and RNA, evidence of DNA misregulation in ALS pathogenesis is relatively scarce. However, emerging research emphasizes DNA damage and defective DNA repair as crucial mechanisms in ALS [25], supported by postmortem studies revealing significant DNA damage in spinal motor neurons and the motor cortex [26,27]. These studies also highlight the activation of DNA damage response (DDR) pathways, including phosphorylated histone H2AX (γ-H2AX), phosphorylated ataxia telangiectasia mutated (p-ATM), and nuclear BRCA1, indicating persistent repair attempts [26,27,28]. TDP-43 and FUS regulate DDR and DNA repair through mechanisms such as non-homologous end joining (NHEJ), homologous recombination (HR), and base excision repair (BER) [25]. Thus, the pathological cytoplasmic mislocalization of TDP-43 and FUS in ALS disrupts nuclear DDR, leading to repair deficiencies [29,30]. C9ORF72 HREs form G-quadruplexes and generate transcription-induced R-loops with RNA [31,32], destabilizing DNA and potentially increasing susceptibility to DNA damage [33]. Additionally, DPRs have been shown to inhibit NHEJ [34].
Despite significant progress over decades of research, the molecular mechanisms underlying ALS pathogenesis remain complex and multifactorial, prompting the question of how current knowledge converges or integrates to explain ALS pathogenesis. To address this challenge, investigating newly identified ALS-associated genes could provide valuable insights into the molecular pathogenesis from a novel perspective. Mutations in NEK1, C21ORF2, and CCNF (encoding Cyclin F) in ALS patients were identified in 2016, and studies conducted by our group and others on these genes have uncovered two novel pathways—the C21ORF2-NEK1 and Cyclin F-VCP pathways—in ALS pathogenesis. This review summarizes the functions of these proteins and explores their potential contributions to a more comprehensive understanding of the molecular mechanisms underlying ALS.
2. NEK1 and C21ORF2
2.1. Human Genetics
Mutations in the NEK1 gene have been associated with a variety of disorders, including short-rib polydactyly syndrome [35], axial spondylometaphyseal dysplasia (SMD) [36], frontotemporal dementia (FTD) with ALS [37], and ALS [37,38,39,40]. A meta-analysis revealed that the prevalence of NEK1 mutations in ALS patients is 3.1% [41]. Likewise, mutations in C21ORF2 (also referred to as CFAP410 or LRRC76) have been identified in patients with SMD [42] and ALS [43]. Furthermore, retinal dystrophy [44,45] and asphyxiating thoracic dystrophy (also known as Jeune syndrome) [46] have also been linked to mutations in C21ORF2. To date, a meta-analysis specifically investigating the prevalence of C21ORF2 mutations in ALS patients has not been conducted.
2.2. Ciliogenesis
All diseases associated with mutations in NEK1 or C21ORF2, with the exception of ALS, are classified as ciliopathies, where defects in ciliogenesis are thought to underlie the pathology [47]. Consequently, the role of NEK1 and C21ORF2 in ciliogenesis has been extensively studied (Figure 1). Gene knockout (KO) of either NEK1 or C21ORF2 in human untransformed retinal pigment epithelial cells results in a significant reduction in ciliogenesis, suggesting that both NEK1 and C21ORF2 are essential for ciliogenesis [48]. At the molecular level, NEK1 and C21ORF2 form a stable complex, mediated by a C-terminal acidic domain in NEK1 (aa 1,160–1,286), referred to as the C21ORF2 Interaction Domain (CID) [48], which facilitates mutual stabilization of the proteins [48,49] (Figure 1).
Pathogenic point mutations in C21ORF2 (R73P and L224P) or in the CID of NEK1 (D1277A) weaken this interaction [48], indicating that complex formation is crucial for their functional roles. Consistently, the NEK1 D1277A mutant is unable to fully rescue ciliogenesis defects in NEK1 KO cells, and C21ORF2 mutants (R73P and L224P) fail to completely restore ciliogenesis in C21ORF2 KO cells. Centrosomes act as nucleation sites for cilia, and both NEK1 and C21ORF2 are localized to these structures [50,51]. ALS-associated C21ORF2 mutants (V58L, R106C, R172W, A255T) exhibit reduced localization to centrosomes and diminished ability to rescue ciliogenesis defects in human neuroblastoma SH-SY5Y cells [52], suggesting these proteins function at the centrosome to initiate ciliogenesis. However, the precise molecular mechanisms by which NEK1 and C21ORF2 regulate ciliogenesis remain to be fully elucidated. Notably, overexpression of NEK1 also inhibits ciliogenesis [51], underscoring the importance of balanced activity in NEK1 for the regulation of ciliogenesis.
2.3. DNA Repair
NEK1 has been identified as playing a critical role in DNA damage repair (Figure 1), a vital defense mechanism for the survival of post-mitotic neurons [53]. The accumulation of DNA damage with aging may serve as a central pathological driver in ALS progression [54]. Upon exposure to DNA-damaging ionizing radiation, NEK1 rapidly localizes to DNA damage sites, positioning itself as an early responder in the DNA damage response (DDR) pathway [55]. NEK1 interacts with key DDR components, including Ataxia Telangiectasia and Rad3-related (ATR) and ATR-interacting protein (ATRIP), thereby priming ATR for efficient DNA damage signaling, which is essential for activation of downstream repair pathways [56].
In addition to its role in DDR signaling, NEK1 directly participates in both NHEJ and HR DNA repair by regulating critical repair factors. NEK1 interacts with Ku80, a key component of the NHEJ pathway, facilitating the loading of replication factors onto chromatin and promoting S-phase progression during DNA replication and repair [57]. NEK1 also regulates RAD54, an essential factor in HR, thus orchestrating HR repair and ensuring replication fork stability [58]. Consistent with this, loss of NEK1 function severely impairs DNA repair capacity, resulting in delayed repair kinetics and failure to properly arrest the cell cycle following DNA damage [59], thereby increasing sensitivity to DNA-damaging agents [60]. NEK1 also increases the expression of DNA repair pathway genes [61]. These findings underscore NEK1 as a multifunctional kinase that integrates DNA damage recognition, repair coordination, and gene regulation to maintain genomic integrity.
Studies involving ALS patient-derived motor neurons have shown that loss of NEK1 function leads to a significant accumulation of DNA damage. Specifically, NEK1 loss-of-function (LoF) mutations result in increased levels of γH2AX foci, a marker of DNA double-strand breaks, and reduced DNA repair capacity in motor neurons [62]. The DNA damage response pathway is notably compromised in these cells, with impaired recruitment of DNA repair proteins to damage sites. Furthermore, in iPSC-derived motor neurons carrying both the HREs in C9ORF72 and a LoF mutation in NEK1, an increase in RNA foci and exacerbation of DNA damage were observed compared to C9ORF72 single mutants. These findings suggest that NEK1 dysfunction exacerbates DNA damage in C9ORF72-mutant motor neurons, potentially acting as a genetic modifier in ALS pathology [63]. The accumulation of DNA damage in motor neurons due to NEK1 dysfunction represents a potential mechanism contributing to motor neuron degeneration in ALS, emphasizing the importance of proper DNA repair mechanisms in maintaining motor neuron health and survival.
C21ORF2 is also implicated in DNA repair processes (Figure 1), as depletion of C21ORF2 reduces the efficiency of HR repair of damaged DNA, which can be rescued by NEK1 overexpression [48,64]. These results suggest that C21ORF2 functions within the same pathway as NEK1 in DNA damage repair, as it does in ciliogenesis. However, in contrast to NEK1, C21ORF2 does not translocate to damaged DNA sites [64], indicating its supportive, rather than essential, role in NEK1 function in DNA repair processes. Recent studies have highlighted that primary cilia play a crucial role in maintaining genome stability by regulating DNA repair processes in cholangiocytes [65]. For instance, DNA repair proteins such as RAD51, ATR, PARP1, CHK1, and CHK2 have been found to colocalize with primary cilia [65]. Additionally, deciliated cells exhibit downregulation of key DDR proteins, such as ATM, p53, and p21, following irradiation, further emphasizing the role of primary cilia in maintaining genomic integrity [65]. Consistently, deciliated cells exhibit reduced survival, increased S-phase arrest, and enhanced DNA damage when exposed to genotoxic agents [65]. While these data suggest that ciliogenesis defects contribute to defective DDR and DNA damage repair, the necessity of primary cilia for DDR and DNA damage repair, along with the detailed mechanisms involved, requires further elucidation in motor neurons.
2.4. Protein Homeostasis
NEK1 is a pivotal kinase that regulates protein stability through the phosphorylation of target proteins (Figure 1). Recent studies have uncovered distinct regulatory mechanisms by which NEK1 influences various substrates. NEK1 directly phosphorylates the Von Hippel-Lindau (VHL) tumor suppressor, promoting its proteasome-dependent degradation [66]. This phosphorylation-induced destabilization of VHL has been shown to affect ciliogenesis and maintenance [66]. In contrast, NEK1 exhibits different regulatory effects on C21ORF2, as described in Section 2.1. NEK1-mediated hyperphosphorylation of C21ORF2 prevents its interaction with the ubiquitin ligase F-box only protein 3 (FBXO3), thereby stabilizing C21ORF2 [49] Notably, the ALS-associated C21ORF2 mutant (V58L) displays heightened susceptibility to NEK1-mediated hyperphosphorylation, suggesting a potential role in ALS pathogenesis [49]. These findings illustrate the bidirectional regulatory capacity of NEK1, which can either promote protein degradation, as seen with VHL, or enhance protein stability, as observed with C21ORF2. This dual regulatory mechanism underscores NEK1’s significance in fine-tuning various cellular functions through the differential control of protein stability. The contrasting effects of NEK1-mediated phosphorylation on different substrates highlight its complex role in cellular homeostasis and disease pathogenesis.
NEK1 is also essential for global protein homeostasis through its interactions with key regulators, including heat shock proteins (HSPA1A, HSPA8, HSP90A1, and HSP90AB1) and ubiquitin [67]. These proteins govern protein folding, repair, and degradation—processes closely linked to neurodegenerative diseases such as ALS. Loss of NEK1 disrupts these pathways, leading to the accumulation of misfolded proteins and impaired clearance [67]. Proteomic analysis of NEK1-deficient motor neurons has revealed significant changes in protein expression, reinforcing its role in proteostasis [67]. Given that protein aggregation and impaired degradation are hallmarks of ALS, NEK1 dysfunction may contribute to disease progression by destabilizing proteostasis networks.
Dysregulation of NEK1-mediated proteostasis may extend beyond its effects on other proteins to impact NEK1 itself, potentially contributing to ALS pathogenesis. Although the evidence is limited, emerging data suggest that, in addition to NEK1 dysfunction, increased NEK1 activity or accumulation may also play a role in ALS. While nonsense mutations in NEK1 primarily support a LoF mechanism, the effects of missense mutations, such as R261H, on NEK1 kinase activity and protein expression remain unclear [38,39]. NEK1 has been shown to phosphorylate C21ORF2, preventing its degradation by the FBXO3-mediated ubiquitin-proteasome system (UPS), as described above. This phosphorylation-induced stabilization of C21ORF2, in turn, enhances NEK1 stability, creating a self-reinforcing regulatory loop in which NEK1 indirectly regulates its own protein levels [49]. Disruption of this cycle, whether through LoF or abnormal stabilization, could contribute to ALS pathogenesis. Consistently, the ALS-associated C21ORF2-V58L variant has been suggested to abnormally stabilize NEK1, leading to its increased accumulation [49]. Notably, in the motor cortex of an ALS patient carrying the R261H missense mutation, cytoplasmic NEK1 protein accumulation have been observed, along with TDP-43 aggregation [68]. These findings raise critical questions about the regulation of NEK1 protein levels in ALS pathogenesis, warranting further investigation.
3. Cyclin F and VCP
3.1. Human Genetics
Point mutations in the exonic regions of the Cyclin F gene (CCNF) have been identified in both familial and sporadic ALS patients, with or without frontotemporal dementia (FTD) [69]. All CCNF mutations in these patients lead to amino acid substitutions, with no deletions or protein-disrupting mutations detected, suggesting a gain-of-function (GoF) mechanism that is toxic to cells [69]. To date, no other diseases associated with CCNF mutations have been identified, although elevated Cyclin F expression levels have been correlated with poorer prognosis in patients with hepatocellular carcinoma [70], clear cell renal cell carcinoma [71], ovarian cancer [72], and breast cancer [73]. The frequency of CCNF mutations in ALS patients is estimated to range from 0.6% to 3.3% [69]. VCP mutations have been implicated in multisystem proteinopathy 1 (MSP1) (also known as inclusion body myopathy associated with Paget disease of bone and FTD) [74,75], Charcot-Marie-Tooth type 2 disease [76], Parkinson’s disease (PD) [77,78], hereditary spastic paraplegia [79], and ALS [80], with the latter condition being added to the list in 2010. Mutations in VCP do not result in deletions, indicating a GoF mechanism, similar to that of CCNF mutations. Studies on patients with VCP mutations report that myopathy, Paget’s disease of bone, and FTD were present in 90%, 42%, and 30% of the patients, respectively, while approximately 9% of patients exhibited an ALS phenotype and 4% were diagnosed with PD [75].
3.2. VCP Activation by Cyciln F
To elucidate the molecular mechanisms underlying ALS pathogenesis, we investigated the potential interaction of Cyclin F with other proteins implicated in ALS and discovered that wild-type Cyclin F binds to p62 (also known as SQSTM1), TDP-43, and VCP, with the strongest binding observed to VCP [83]. VCP functions as a molecular chaperone and segregase, utilizing ATP hydrolysis to extract proteins from large cellular structures for degradation or recycling, thereby maintaining protein homeostasis and supporting various cellular processes [84,85]. We found that the N-terminal region of Cyclin F is responsible for its interaction with VCP (Figure 2), and mutations identified in ALS patients enhance this binding. In parallel with this binding, the ATPase activity of VCP is significantly increased [83]. Furthermore, some of the mutations mislocalize Cyclin F to the cytoplasm, whereas the wild-type protein predominantly resides in the nucleus [83].
As described earlier, VCP itself is subject to point mutations in ALS [80], some of which have been shown to augment its ATPase activity [86,87]. Given that VCP is reported to possess unfoldase activity mediated by its ATPase function [88], and that protein aggregation, frequently observed in ALS patient motor neurons, is thought to be driven by unstructured regions susceptible to unfolding [89], we hypothesized that Cyclin F mutants, which activate VCP’s ATPase activity, lead to excessive unfoldase activity of VCP, resulting in abnormal protein unfolding and subsequent aggregation. Consistent with this hypothesis, we demonstrated that increased VCP ATPase activity promotes oxidative stress-induced TDP-43 cytoplasmic aggregation [83], a characteristic observed in approximately 90% of ALS patients [90]. In support of our findings, a recent study highlighted the critical role of molecular chaperones in TDP-43 folding, which prevents the formation of TDP-43 aggregates [91,92,93]. Furthermore, recent experiments in which wild-type and mutant Cyclin F were overexpressed in the mouse brain demonstrated that mutant Cyclin F promotes TDP-43 aggregation with colocalization of VCP [94], further corroborating our results.
Although Cyclin F is a prototypical F-box protein that functions as a substrate receptor in the Cullin1-Ring ubiquitin ligase complex (CRL1, also known as SCF) [95], we did not observe changes in the ubiquitylation levels of VCP in cells overexpressing Cyclin F [83], despite extensive ubiquitylation of the known substrate, RRM2, in the same experimental condition [83,96]. Therefore, ubiquitylation likely does not contribute to Cyclin F-mediated activation of VCP ATPase activity, revealing a novel function of Cyclin F.
3.3. Cyclin F as an Ubiquitin Ligase for ALS-Associated Proteins
While our study primarily focused on VCP among the Cyclin F binding proteins, other research has investigated the direct interaction between Cyclin F and TDP-43, reporting that Cyclin F directly binds to TDP-43 and facilitates CRL1Cyclin F-mediated ubiquitylation, leading to proteasomal degradation [97,98]. Interestingly, Cyclin F mutants derived from ALS patients were shown to enhance K48-linked poly ubiquitylation, a well-established marker for proteasomal degradation [99], yet instead of promoting degradation, these mutants result in protein aggregation [97]. This discrepancy warrants further investigation. Additionally, while these findings are intriguing, they require validation under more physiological conditions, particularly since the data were primarily derived from experiments involving overexpressed Cyclin F and TDP-43. Notably, Cyclin F is an unstable protein, whereas TDP-43 is relatively stable, suggesting that the effects of Cyclin F on TDP-43 may be transient. Furthermore, the overexpression of Cyclin F could introduce experimental artifacts.
In addition to TDP-43, p62 has also been reported to undergo ubiquitylation by CRL1Cyclin F, promoting p62 aggregation [100]. Compared to wild-type Cyclin F, the ALS patient-derived Cyclin F mutant exhibits a greater ability to enhance p62 ubiquitylation but unexpectedly reduces p62 aggregation [100]. Thus, no direct correlation between ubiquitylation and p62 aggregation is evident. Similar to TDP-43, this study primarily relied on protein overexpression, and therefore, the results require validation in more physiologically relevant contexts. Importantly, p62 has been shown to enhance TDP-43 aggregation [101], suggesting that Cyclin F, VCP, and p62 may act synergistically to promote the formation of pathogenic TDP-43 aggregates.
4. Possible Convergent Mechanisms of ALS
The discussions in the preceding sections suggest that mutations in C21ORF2, NEK1, Cyclin F, and VCP play central roles in ALS pathogenesis. C21ORF2 mutant-induced stabilization of NEK1, together with Cyclin F mutant-induced VCP activation and the subsequent aggregation of TDP-43, provides compelling evidence that multiple pathways contribute to ALS. However, the question remains whether these mechanisms operate independently or converge at a critical juncture in the disease process. TDP-43 aggregation is a hallmark of ALS, and its presence has been noted in ALS patients with NEK1 mutations [68]. This observation raises the possibility that TDP-43 aggregation might occur at or downstream of an intersection between these pathways. Despite such evidence, the exact mechanism by which NEK1 mutations induce TDP-43 aggregation remains unclear, suggesting the need for further investigation to uncover whether these processes are truly independent or intertwined.
The Cyclin F T31 phosphorylation site, which is catalyzed by AKT [102], lies within a preferred sequence for NEK1 phosphorylation [103], suggesting the intersection between Cyclin F and NEK1. NEK1’s potential role in phosphorylating this site may serve to stabilize Cyclin F, enhancing VCP ATPase activity and promoting subsequent TDP-43 aggregation, which is a central pathological feature in ALS. TDP-43 aggregation is now understood to mediate ALS pathogenesis through both LoF and GoF mechanisms [104,105]. The aggregation of TDP-43 impairs its normal role in regulating mRNA splicing, a critical function for the maintenance of cellular homeostasis. Additionally, it leads to the sequestration of essential proteins that govern proteostasis and protein transport [106,107]. The disruption of these cellular processes is one of the core drivers of ALS progression. Therefore, understanding the precise factors contributing to TDP-43 aggregation—such as the potential convergence of NEK1 and Cyclin F signaling—could provide valuable insights into ALS pathogenesis and novel therapeutic targets.
In addition to its role in mRNA processing, emerging evidence supports a critical role for TDP-43 in DNA damage repair (Figure 3). TDP-43 translocates to DNA damage sites upon double-strand breaks, where it recruits DNA ligase to seal the breaks, facilitating the repair process [30]. In instances of DNA mismatch, TDP-43 interacts with mismatch repair proteins such as MLH1 and MSH6, which are crucial for efficient repair [108]. Furthermore, TDP-43 has been shown to regulate the expression of these mismatch repair proteins even in the absence of DNA-damaging stress, suggesting that TDP-43 plays an important role in enhancing cellular resistance to DNA damage [109]. This adds another layer of complexity to the role of TDP-43 in ALS, as disruption of DNA repair mechanisms may contribute to the pathogenesis of the disease. The pathological aggregation of TDP-43 in the cytoplasm disrupts nuclear transport by displacing transport factors from their proper localization, as described earlier [106]. This disruption affects the nuclear translocation of proteins involved in DNA damage repair, thereby potentially compromising genomic integrity. The ALS-associated TDP-43 Q331K mutant, which exhibits reduced nuclear localization and increased cytoplasmic mislocalization, impairs the nuclear import of XRCC4-DNA ligase 4, a critical component of the NHEJ pathway [110]. Furthermore, ALS/FTD patients with TDP-43 pathology exhibit an accumulation of the DNA damage markers, further supporting the hypothesis that TDP-43 dysfunction leads to impaired DNA repair and genomic instability [26,27,28]. Collectively, these findings suggest that defects in TDP-43-mediated DNA repair mechanisms contribute to disease pathogenesis, potentially exacerbating neuronal vulnerability in ALS.
Interestingly, in addition to NEK1/C21ORF2 module, cyclin F and VCP have also been implicated in DNA damage repair. Cyclin F, as part of the CRL1 ubiquitin ligase complex, targets atypical E2F transcription factors (E2F7 and E2F8) for proteasomal degradation [111]. This degradation is essential for the expression of DNA repair genes which are involved in various DNA repair pathways [111]. Cyclin F also contributes to maintaining genome integrity by regulating degradation of RRM2, a regulatory component of dNTP-generating ribonucleotide reductase complex [96]. Cyclin F is downregulated in response to DNA damage to allow accumulation of RRM2 and support DNA damage repair [96]. VCP is involved in the mobilization of damage sensors to repair executors by extracting these sensors from chromatin using its ATPase activity [112,113]. This mechanism is essential for nucleotide excision repair and double-strand break repair [112,113]. Given the essential role of VCP in mobilizing repair proteins, it is likely that mutations in VCP or alterations in its regulation by Cyclin F could lead to impaired DNA repair and contribute to ALS pathogenesis (Figure 3).
Considering the substantial accumulation of DNA damage observed in the motor neurons of ALS patients [26,27,28], it is plausible that impaired DNA damage repair represents a common downstream mechanism in ALS pathogenesis. Mutations in C21ORF2, NEK1, Cyclin F, and VCP compromise DNA repair pathways, leading to genomic instability. As DNA repair mechanisms are disrupted, the accumulation of genome mutations might cause the accumulation of abnormal proteins which may disrupt the protein homeostasis and accelerate neurodegeneration, contributing to the onset and progression of ALS (Figure 3). Therefore, understanding how these mutations converge to impair DNA repair, protein homeostasis, and TDP-43 function could provide critical insights into the molecular underpinnings of ALS and open new avenues for therapeutic intervention.
Author Contributions
Conceptualization, Y.W. and T.N.; investigation, Y.W., T.N. and M.N.; writing—original draft preparation, Y.W. and T.N.; writing—review and editing, Y.W., T.N. and K.N.; supervision, T.N. and K.N.; project administration, T.N. and K.N.; funding acquisition, T.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by KAKENHI (grant number: 23K06367).
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
We would like to thank lab members for the productive discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brown, R.H. and A. Al-Chalabi, Amyotrophic Lateral Sclerosis. N Engl J Med, 2017. 377(2): p. 162-172.
- Wang, H., L. Guan, and M. Deng, Recent progress of the genetics of amyotrophic lateral sclerosis and challenges of gene therapy. Front Neurosci, 2023. 17: p. 1170996.
- Mead, R.J. , et al., Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov, 2023. 22(3): p. 185-212.
- Soto, C. and S. Pritzkow, Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci, 2018. 21(10): p. 1332-1340.
- Moda, F. , et al., Secondary Protein Aggregates in Neurodegenerative Diseases: Almost the Rule Rather than the Exception. Front Biosci (Landmark Ed), 2023. 28(10): p. 255.
- Menzies, F.M. , et al., Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain, 2002. 125(Pt 7): p. 1522-33.
- Pedrini, S. , et al., ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet, 2010. 19(15): p. 2974-86.
- Richardson, K. , et al., The effect of SOD1 mutation on cellular bioenergetic profile and viability in response to oxidative stress and influence of mutation-type. PLoS One, 2013. 8(6): p. e68256.
- Ash, P.E. , et al., Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron, 2013. 77(4): p. 639-46.
- Lee, K.H. , et al., C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell, 2016. 167(3): p. 774-788.e17.
- Ryan, S. , et al., C9orf72 dipeptides disrupt the nucleocytoplasmic transport machinery and cause TDP-43 mislocalisation to the cytoplasm. Sci Rep, 2022. 12(1): p. 4799.
- Johnson, B.S. , et al., TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem, 2009. 284(30): p. 20329-39.
- Nonaka, T. , et al., Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet, 2009. 18(18): p. 3353-64.
- Dormann, D. , et al., ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. Embo j, 2010. 29(16): p. 2841-57.
- Kino, Y. , et al., Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res, 2011. 39(7): p. 2781-98.
- Ederle, H. and D. Dormann, TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett, 2017. 591(11): p. 1489-1507.
- Chen, H.J. , et al., The heat shock response plays an important role in TDP-43 clearance: evidence for dysfunction in amyotrophic lateral sclerosis. Brain, 2016. 139(Pt 5): p. 1417-32.
- Riemenschneider, H. , et al., Gel-like inclusions of C-terminal fragments of TDP-43 sequester stalled proteasomes in neurons. EMBO Rep, 2022. 23(6): p. e53890.
- Zhao, M. , et al., RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol Cells, 2018. 41(9): p. 818-829.
- Xue, Y.C. , et al., Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front Mol Neurosci, 2020. 13: p. 78.
- Sareen, D. , et al., Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med, 2013. 5(208): p. 208ra149.
- Lagier-Tourenne, C. , et al., Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A, 2013. 110(47): p. E4530-9.
- Zu, T. , et al., RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci U S A, 2013. 110(51): p. E4968-77.
- Tran, H. , et al., Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron, 2015. 87(6): p. 1207-1214.
- Sun, Y. , et al., The role of DNA damage response in amyotrophic lateral sclerosis. Essays Biochem, 2020. 64(5): p. 847-861.
- Farg, M.A. , et al., The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet, 2017. 26(15): p. 2882-2896.
- Kim, B.W. , et al., DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol Commun, 2020. 8(1): p. 7.
- Fang, M. , et al., Loss of TDP-43 function contributes to genomic instability in amyotrophic lateral sclerosis. Front Neurosci, 2023. 17: p. 1251228.
- Wang, H. , et al., Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat Commun, 2018. 9(1): p. 3683.
- Mitra, J. , et al., Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A, 2019. 116(10): p. 4696-4705.
- Reddy, K. , et al., The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J Biol Chem, 2013. 288(14): p. 9860-9866.
- Haeusler, A.R. , et al., C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature, 2014. 507(7491): p. 195-200.
- Crossley, M.P., M. Bocek, and K.A. Cimprich, R-Loops as Cellular Regulators and Genomic Threats. Mol Cell, 2019. 73(3): p. 398-411.
- Andrade, N.S. , et al., Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol Neurodegener, 2020. 15(1): p. 13.
- Thiel, C. , et al., NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet, 2011. 88(1): p. 106-14.
- Wang, Z. , et al., Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J Hum Genet, 2017. 62(4): p. 503-506.
- Nguyen, H.P. , et al., NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol Aging, 2018. 61: p. 255.e1-255.e7.
- Brenner, D. , et al., NEK1 mutations in familial amyotrophic lateral sclerosis, in Brain. 2016: England. p. e28.
- Kenna, K.P. , et al., NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet, 2016. 48(9): p. 1037-42.
- Gratten, J. , et al., Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med, 2017. 9(1): p. 97.
- Yao, L. , et al., NEK1 mutations and the risk of amyotrophic lateral sclerosis (ALS): a meta-analysis. Neurol Sci, 2021. 42(4): p. 1277-1285.
- Wang, Z. , et al., Axial Spondylometaphyseal Dysplasia Is Caused by C21orf2 Mutations. PLoS One, 2016. 11(3): p. e0150555.
- van Rheenen, W. , et al., Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet, 2016. 48(9): p. 1043-8.
- Khan, A.O. , et al., C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br J Ophthalmol, 2015. 99(12): p. 1725-31.
- Suga, A. , et al., Identification of Novel Mutations in the LRR-Cap Domain of C21orf2 in Japanese Patients With Retinitis Pigmentosa and Cone-Rod Dystrophy. Invest Ophthalmol Vis Sci, 2016. 57(10): p. 4255-63.
- Wheway, G. , et al., An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat Cell Biol, 2015. 17(8): p. 1074-1087.
- Reiter, J.F. and M.R. Leroux, Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol, 2017. 18(9): p. 533-547.
- Gregorczyk, M. , et al., Functional characterization of C21ORF2 association with the NEK1 kinase mutated in human in diseases. Life Sci Alliance, 2023. 6(7).
- Watanabe, Y. , et al., An Amyotrophic Lateral Sclerosis-Associated Mutant of C21ORF2 Is Stabilized by NEK1-Mediated Hyperphosphorylation and the Inability to Bind FBXO3. iScience, 2020. 23(9): p. 101491.
- Mahjoub, M.R., M. L. Trapp, and L.M. Quarmby, NIMA-related kinases defective in murine models of polycystic kidney diseases localize to primary cilia and centrosomes. J Am Soc Nephrol, 2005. 16(12): p. 3485-9.
- White, M.C. and L.M. Quarmby, The NIMA-family kinase, Nek1 affects the stability of centrosomes and ciliogenesis. BMC Cell Biol, 2008. 9: p. 29.
- De Decker, M. , et al., C21ORF2 mutations point towards primary cilia dysfunction in amyotrophic lateral sclerosis. Brain, 2025. 148(3): p. 803-816.
- Delint-Ramirez, I. and R. Madabhushi, DNA damage and its links to neuronal aging and degeneration. Neuron, 2025. 113(1): p. 7-28.
- Wang, H. , et al., DNA Damage and Repair Deficiency in ALS/FTD-Associated Neurodegeneration: From Molecular Mechanisms to Therapeutic Implication. Front Mol Neurosci, 2021. 14: p. 784361.
- Polci, R. , et al., NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res, 2004. 64(24): p. 8800-3.
- Liu, S. , et al., Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc Natl Acad Sci U S A, 2013. 110(6): p. 2175-80.
- Patil, M. , et al., Nek1 interacts with Ku80 to assist chromatin loading of replication factors and S-phase progression. Cell Cycle, 2013. 12(16): p. 2608-16.
- Spies, J. , et al., Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol Cell, 2016. 62(6): p. 903-917.
- Pelegrini, A.L. , et al., Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis, 2010. 25(5): p. 447-54.
- Melo-Hanchuk, T.D. , et al., NEK1 kinase domain structure and its dynamic protein interactome after exposure to Cisplatin. Sci Rep, 2017. 7(1): p. 5445.
- Martins, M.B. , et al., NEK1 deficiency affects mitochondrial functions and the transcriptome of key DNA repair pathways. Mutagenesis, 2021. 36(3): p. 223-236.
- Higelin, J. , et al., NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res, 2018. 30: p. 150-162.
- Santangelo, S. , et al., NEK1 haploinsufficiency worsens DNA damage, but not defective ciliogenesis, in C9ORF72 patient-derived iPSC-motoneurons. Hum Mol Genet, 2024. 33(21): p. 1900-1907.
- Fang, X. , et al., The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim Biophys Sin (Shanghai), 2015. 47(10): p. 834-41.
- Peixoto, E. , et al., Cholangiocytes’ Primary Cilia Regulate DNA Damage Response and Repair. bioRxiv, 2025.
- Patil, M. , et al., Nek1 phosphorylates Von Hippel-Lindau tumor suppressor to promote its proteasomal degradation and ciliary destabilization. Cell Cycle, 2013. 12(1): p. 166-71.
- Mann, J.R. , et al., Loss of function of the ALS-associated NEK1 kinase disrupts microtubule homeostasis and nuclear import. Sci Adv, 2023. 9(33): p. eadi5548.
- Rifai, O.M. , et al., Clinicopathological analysis of NEK1 variants in amyotrophic lateral sclerosis. Brain Pathol, 2025. 35(1): p. e13287.
- Williams, K.L. , et al., CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun, 2016. 7: p. 11253.
- Zelong, Y. , et al., Increased expression of Cyclin F in liver cancer predicts poor prognosis: A study based on TCGA database. Medicine (Baltimore), 2021. 100(31): p. e26623.
- Kwiatkowski, M. , et al., Overexpression of cyclin F/CCNF as an independent prognostic factor for poor survival in clear cell renal cell carcinoma. Sci Rep, 2024. 14(1): p. 9280.
- Li, Y. , et al., Cyclin F and KIF20A, FOXM1 target genes, increase proliferation and invasion of ovarian cancer cells. Exp Cell Res, 2020. 395(2): p. 112212.
- Liu, Y. , et al., Systematic analysis of the expression and prognosis relevance of FBXO family reveals the significance of FBXO1 in human breast cancer. Cancer Cell Int, 2021. 21(1): p. 130.
- Watts, G.D. , et al., Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet, 2004. 36(4): p. 377-81.
- Al-Obeidi, E. , et al., Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet, 2018. 93(1): p. 119-125.
- Gonzalez, M.A. , et al., A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain, 2014. 137(Pt 11): p. 2897-902.
- Chan, N. , et al., Valosin-containing protein mutation and Parkinson’s disease, in Parkinsonism Relat Disord. 2012: England. p. 107-9.
- Majounie, E. , et al., Mutational analysis of the VCP gene in Parkinson’s disease. Neurobiol Aging, 2012. 33(1): p. 209.e1-2.
- van de Warrenburg, B.P. , et al., Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur J Hum Genet, 2016. 24(10): p. 1460-6.
- Johnson, J.O. , et al., Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron, 2010. 68(5): p. 857-64.
- Rayner, S.L. , et al., Cyclin F, Neurodegeneration, and the Pathogenesis of ALS/FTD. Neuroscientist, 2024. 30(2): p. 214-228.
- Scarian, E. , et al., The Role of VCP Mutations in the Spectrum of Amyotrophic Lateral Sclerosis-Frontotemporal Dementia. Front Neurol, 2022. 13: p. 841394.
- Yu, Y. , et al., Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum Mol Genet, 2019. 28(20): p. 3486-3497.
- Wang, Q., C. Song, and C.C. Li, Molecular perspectives on p97-VCP: progress in understanding its structure and diverse biological functions. J Struct Biol, 2004. 146(1-2): p. 44-57.
- Braxton, J.R. and D.R. Southworth, Structural insights of the p97/VCP AAA+ ATPase: How adapter interactions coordinate diverse cellular functionality. J Biol Chem, 2023. 299(11): p. 105182.
- Manno, A. , et al., Enhanced ATPase activities as a primary defect of mutant valosin-containing proteins that cause inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Genes Cells, 2010. 15(8): p. 911-22.
- Rijal, R. , et al., Mutant p97 exhibits species-specific changes of its ATPase activity and compromises the UBXD9-mediated monomerisation of p97 hexamers. Eur J Cell Biol, 2016. 95(6-7): p. 195-207.
- van den Boom, J. and H. Meyer, VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell, 2018. 69(2): p. 182-194.
- Ayyadevara, S. , et al., Intrinsically disordered proteins identified in the aggregate proteome serve as biomarkers of neurodegeneration. Metab Brain Dis, 2022. 37(1): p. 147-152.
- Ling, S.C., M. Polymenidou, and D.W. Cleveland, Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron, 2013. 79(3): p. 416-38.
- Kitamura, A., N. Iwasaki, and M. Kinjo, Molecular chaperone HSP70 prevents formation of inclusion bodies of the 25-kDa C-terminal fragment of TDP-43 by preventing aggregate accumulation. Cell Stress Chaperones, 2018. 23(6): p. 1177-1183.
- Lin, L.T. , et al., Hsp90 and its co-chaperone Sti1 control TDP-43 misfolding and toxicity. Faseb j, 2021. 35(5): p. e21594.
- Lam, A.Y.W. , et al., DNAJA2 and Hero11 mediate similar conformational extension and aggregation suppression of TDP-43. Rna, 2024. 30(11): p. 1422-1436.
- van Hummel, A. , et al., TDP-43 pathology and functional deficits in wild-type and ALS/FTD mutant cyclin F mouse models. Neuropathol Appl Neurobiol, 2023. 49(2): p. e12902.
- Bai, C. , et al., SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell, 1996. 86(2): p. 263-74.
- D’Angiolella, V. , et al., Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell, 2012. 149(5): p. 1023-34.
- Rayner, S.L. , et al., TDP-43 is a ubiquitylation substrate of the SCF(cyclin F) complex. Neurobiol Dis, 2022. 167: p. 105673.
- Rayner, S.L. , et al., Cyclin F can alter the turnover of TDP-43. Neurobiol Dis, 2024. 192: p. 106421.
- Swatek, K.N. and D. Komander, Ubiquitin modifications. Cell Res, 2016. 26(4): p. 399-422.
- Davidson, J.M. , et al., The E3 Ubiquitin Ligase SCF Cyclin F Promotes Sequestosome-1/p62 Insolubility and Foci Formation and is Dysregulated in ALS and FTD Pathogenesis. Mol Neurobiol, 2023. 60(9): p. 5034-5054.
- Foster, A.D. , et al., p62 overexpression induces TDP-43 cytoplasmic mislocalisation, aggregation and cleavage and neuronal death. Sci Rep, 2021. 11(1): p. 11474.
- Choudhury, R. , et al., The E3 Ubiquitin Ligase SCF(Cyclin F) Transmits AKT Signaling to the Cell-Cycle Machinery. Cell Rep, 2017. 20(13): p. 3212-3222.
- van de Kooij, B. , et al., Comprehensive substrate specificity profiling of the human Nek kinome reveals unexpected signaling outputs. Elife, 2019. 8.
- Cascella, R. , et al., Quantification of the Relative Contributions of Loss-of-function and Gain-of-function Mechanisms in TAR DNA-binding Protein 43 (TDP-43) Proteinopathies. J Biol Chem, 2016. 291(37): p. 19437-48.
- Kim, G. , et al., ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron, 2020. 108(5): p. 822-842.
- Chou, C.C. , et al., TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci, 2018. 21(2): p. 228-239.
- Tsekrekou, M. , et al., Protein aggregation and therapeutic strategies in SOD1- and TDP-43- linked ALS. Front Mol Biosci, 2024. 11: p. 1383453.
- Provasek, V.E. , et al., TDP43 interacts with MLH1 and MSH6 proteins in a DNA damage-inducible manner. Mol Brain, 2024. 17(1): p. 32.
- Provasek, V.E. , et al., RNA/DNA Binding Protein TDP43 Regulates DNA Mismatch Repair Genes with Implications for Genome Stability. bioRxiv, 2024.
- Guerrero, E.N. , et al., Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum Mol Genet, 2019. 28(15): p. 2459-2476.
- Yuan, R. , et al., Cyclin F-dependent degradation of E2F7 is critical for DNA repair and G2-phase progression. Embo j, 2019. 38(20): p. e101430.
- Jiang, N. , et al., Valosin-containing protein regulates the proteasome-mediated degradation of DNA-PKcs in glioma cells. Cell Death Dis, 2013. 4(5): p. e647.
- He, J. , et al., Valosin-containing Protein (VCP)/p97 Segregase Mediates Proteolytic Processing of Cockayne Syndrome Group B (CSB) in Damaged Chromatin. J Biol Chem, 2016. 291(14): p. 7396-408.
Figure 1.
(a) Schematic depiction of the domain structures of NEK1 and C21ORF2. NEK1 comprises kinase, basic, and coiled-coil domains, along with nuclear export signals (NES), while C21ORF2 contains leucine-rich repeats (LRR) and an LRR C-terminal flanking region (LRRCT). The C21ORF2-interacting domain (CID) in NEK1 facilitates its interaction with C21ORF2. (b) Cellular processes regulated by the NEK1/C21ORF2 complex. The NEK1/C21ORF2 complex plays a pivotal role in ciliogenesis, DNA repair, and protein homeostasis. The wrench represents proteins that are involved in DNA repair processes.
Figure 1.
(a) Schematic depiction of the domain structures of NEK1 and C21ORF2. NEK1 comprises kinase, basic, and coiled-coil domains, along with nuclear export signals (NES), while C21ORF2 contains leucine-rich repeats (LRR) and an LRR C-terminal flanking region (LRRCT). The C21ORF2-interacting domain (CID) in NEK1 facilitates its interaction with C21ORF2. (b) Cellular processes regulated by the NEK1/C21ORF2 complex. The NEK1/C21ORF2 complex plays a pivotal role in ciliogenesis, DNA repair, and protein homeostasis. The wrench represents proteins that are involved in DNA repair processes.
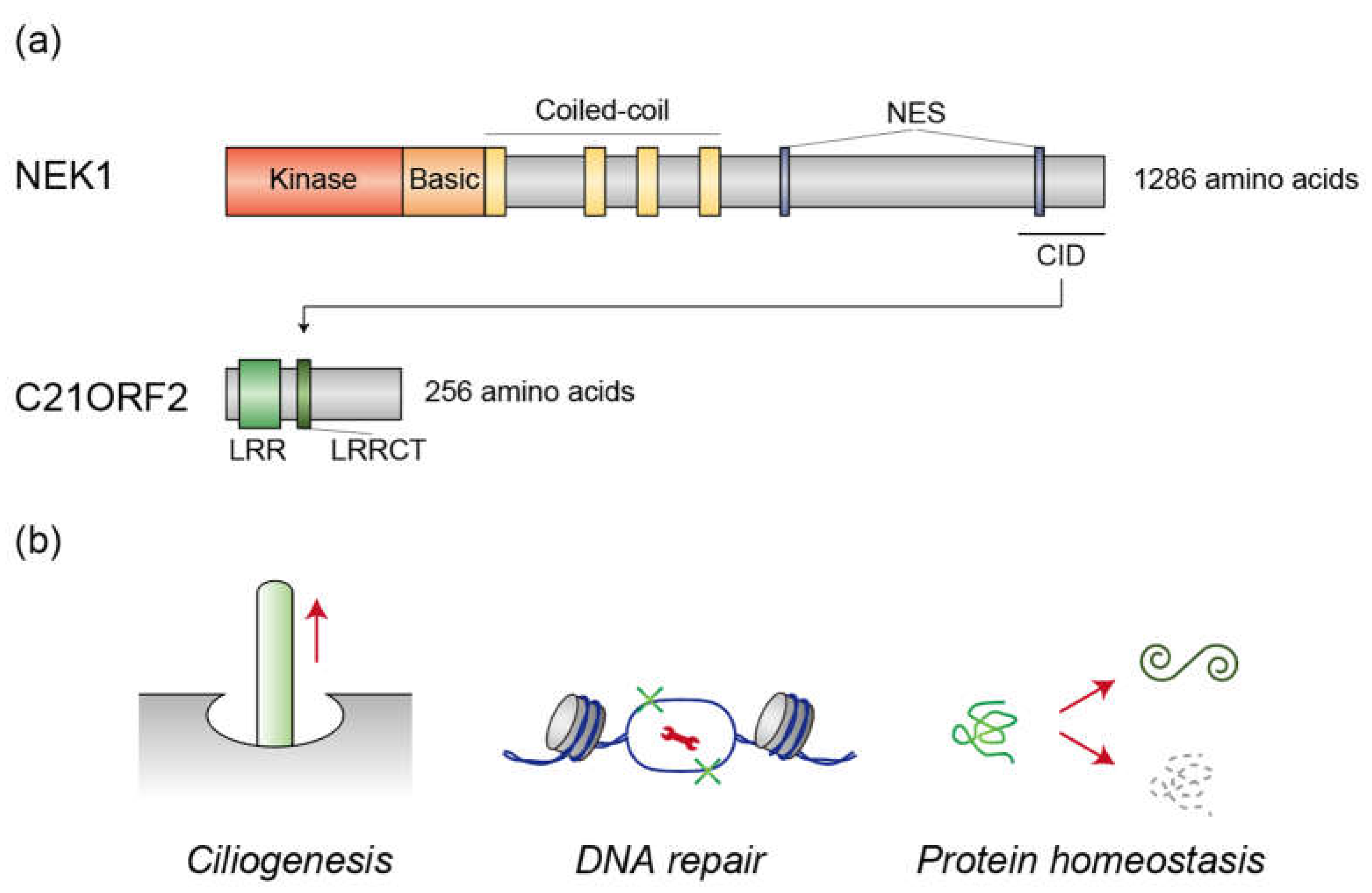
Figure 2.
(a) Schematic representation of the domain structures of Cyclin F and VCP. Cyclin F consists of F-box (F), cyclin, and PEST domains, along with nuclear localization signals (NLS), whereas VCP contains N-terminal and ATPase domains. The N-terminal region of Cyclin F interacts with the ATPase domain of VCP. (b) Molecular mechanism underlying TDP-43 aggregation regulated by Cyclin F and VCP. Cyclin F binds to TDP-43 to facilitate its ubiquitylation by the CUL1 complex. Concurrently, Cyclin F associates with VCP to activate its ATPase activity. VCP induces a conformational change in TDP-43, thereby promoting its aggregation. VCP is known to recognize ubiquitylated proteins, suggesting that TDP-43 ubiquitylation likely precedes recognition by VCP, although it is also possible that TDP-43 is ubiquitylated subsequent to VCP-mediated unfolding.
Figure 2.
(a) Schematic representation of the domain structures of Cyclin F and VCP. Cyclin F consists of F-box (F), cyclin, and PEST domains, along with nuclear localization signals (NLS), whereas VCP contains N-terminal and ATPase domains. The N-terminal region of Cyclin F interacts with the ATPase domain of VCP. (b) Molecular mechanism underlying TDP-43 aggregation regulated by Cyclin F and VCP. Cyclin F binds to TDP-43 to facilitate its ubiquitylation by the CUL1 complex. Concurrently, Cyclin F associates with VCP to activate its ATPase activity. VCP induces a conformational change in TDP-43, thereby promoting its aggregation. VCP is known to recognize ubiquitylated proteins, suggesting that TDP-43 ubiquitylation likely precedes recognition by VCP, although it is also possible that TDP-43 is ubiquitylated subsequent to VCP-mediated unfolding.
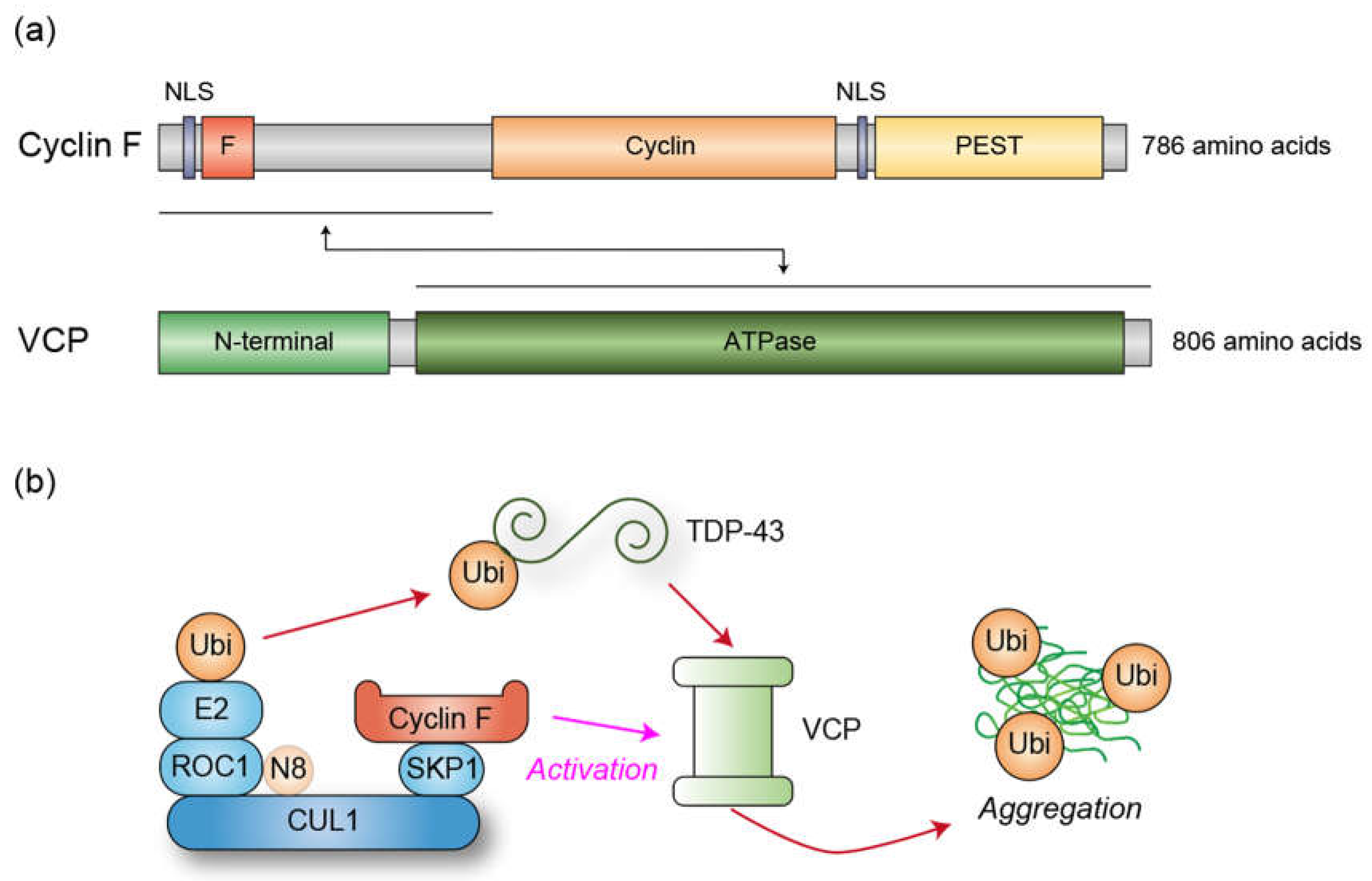
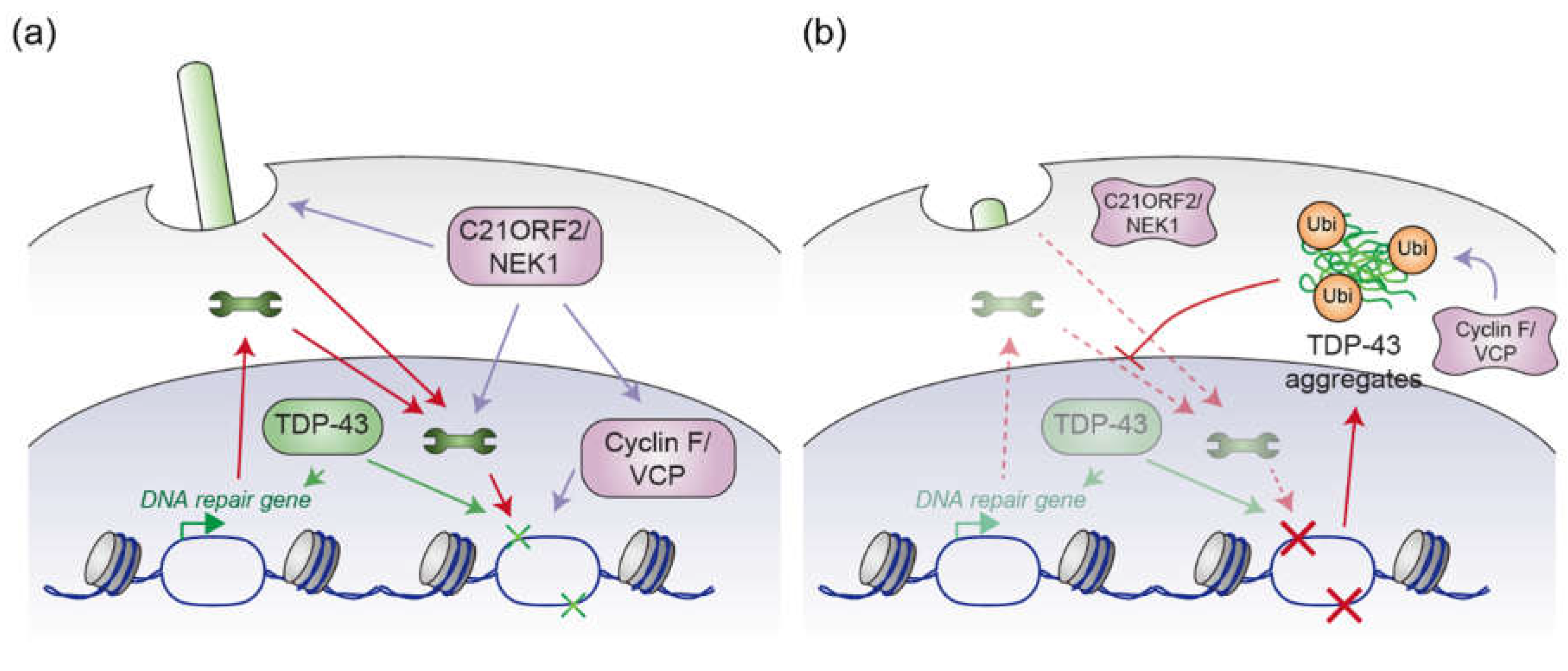
Figure 3.
(a) Potential intersection of the C21ORF2/NEK1 module, the Cyclin F/VCP module, and TDP-43 in healthy motor neurons. The C21ORF2/NEK1 module facilitates ciliogenesis, which has been demonstrated to stabilize proteins involved in DNA repair. Additionally, C21ORF2/NEK1 plays a pivotal role in the activity of DNA repair-associated proteins. NEK1 may also stabilize Cyclin F by phosphorylating T31 residue. VCP, whose activity is supported by Cyclin F, also contributes to DNA repair processes. Furthermore, TDP-43 is implicated in the regulation of genes associated with DNA repair, as well as in the DNA repair mechanisms themselves. (b) Potential intersection of mutant C21ORF2/NEK1, mutant Cyclin F/VCP, and TDP-43 aggregation in the pathogenesis of ALS. Mutations in C21ORF2 affect the protein levels of NEK1 and vice versa, resulting in abnormal NEK1 kinase activity, defects in ciliogenesis, and impairments in DNA repair processes. Mutant Cyclin F, often associated with cytoplasmic translocation, aberrantly activates VCP ATPase activity. This activation, also induced by VCP mutations, promotes the aggregation of TDP-43, as illustrated in Figure 2. These aggregates disrupt nuclear protein import by sequestering proteins involved in nucleocytoplasmic transport, thereby potentially decreasing the concentration of DNA repair-related proteins in the nucleus and impairing cellular DNA repair activity. Simultaneously, this disrupts the ability to enhance the expression of DNA repair genes and the DNA repair processes themselves, due to the reduction of nuclear TDP-43. The failure to repair damaged DNA may lead to genomic mutations that perturb protein homeostasis, promoting the further accumulation of TDP-43 aggregates. The wrench represents the protein that is involved in DNA repair processes.
Figure 3.
(a) Potential intersection of the C21ORF2/NEK1 module, the Cyclin F/VCP module, and TDP-43 in healthy motor neurons. The C21ORF2/NEK1 module facilitates ciliogenesis, which has been demonstrated to stabilize proteins involved in DNA repair. Additionally, C21ORF2/NEK1 plays a pivotal role in the activity of DNA repair-associated proteins. NEK1 may also stabilize Cyclin F by phosphorylating T31 residue. VCP, whose activity is supported by Cyclin F, also contributes to DNA repair processes. Furthermore, TDP-43 is implicated in the regulation of genes associated with DNA repair, as well as in the DNA repair mechanisms themselves. (b) Potential intersection of mutant C21ORF2/NEK1, mutant Cyclin F/VCP, and TDP-43 aggregation in the pathogenesis of ALS. Mutations in C21ORF2 affect the protein levels of NEK1 and vice versa, resulting in abnormal NEK1 kinase activity, defects in ciliogenesis, and impairments in DNA repair processes. Mutant Cyclin F, often associated with cytoplasmic translocation, aberrantly activates VCP ATPase activity. This activation, also induced by VCP mutations, promotes the aggregation of TDP-43, as illustrated in Figure 2. These aggregates disrupt nuclear protein import by sequestering proteins involved in nucleocytoplasmic transport, thereby potentially decreasing the concentration of DNA repair-related proteins in the nucleus and impairing cellular DNA repair activity. Simultaneously, this disrupts the ability to enhance the expression of DNA repair genes and the DNA repair processes themselves, due to the reduction of nuclear TDP-43. The failure to repair damaged DNA may lead to genomic mutations that perturb protein homeostasis, promoting the further accumulation of TDP-43 aggregates. The wrench represents the protein that is involved in DNA repair processes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.
Downloads
63
Views
44
Comments
0
Subscription
Notify me about updates to this article or when a peer-reviewed version is published.




MDPI Initiatives
Important Links
© 2025 MDPI (Basel, Switzerland) unless otherwise stated