
Submitted:
21 March 2025
Posted:
24 March 2025
You are already at the latest version
Abstract
In this study, a Brönsted-Lewis bifunctional acidic catalyst PW/UiO/CNTs-OH was synthesized via hydrothermal method. The esterification reaction parameters between oleic acid and methanol catalyzed by PW/UiO/CNTs-OH were optimized using central composite design-response surface methodology (CCD-RSM). The process achieved 92.9% biodiesel yield under optimized reaction conditions, retaining 82.3% biodiesel yield after four catalytic cycles. The enhanced catalytic performance of PW/UiO/CNTs-OH can be attributed as follows: the [Zr6O4(OH)4]12+ anchored on the surface of MWCNTs provides nucleation sites of UiO-66, enabling dual functions of HPW stabilization and Lewis acid site generation via quadrupole inversion. In addition, HPW introduction during synthesis of UiO-66 reduces solution pH, inducing the protonation of the p-Phthalic acid (PTA) to disrupt the coordination with the [Zr6O4(OH)4] cluster, thereby creating an unsaturated Zr4+ site with electron pair-accepting capability as additional Lewis acid sites. EIS analysis revealed that PW/UiO/CNTs-OH exhibited superior electron migration efficiency compared to UiO-66 and PW/UiO. Furthermore, NH3-TPD and Py-IR analysis showed that PW/UiO/CNTs-OH possessed high densities of Lewis acidic sites of 83.69 μmol/g and Brönsted acidic sites of 9.98 μmol/g.
Keywords:
Biodiesel
; PW/UiO/CNTs-OH
; Brönsted-Lewis dual acidic catalyst
; Response surface methodology (RSM)
1. Introduction
Biodiesel is a fatty acid methyl ester (FAME) produced by the esterification or transesterification of free fatty acids (FFAs) or triglycerides with methanol [1,2]. The process requires catalysts that simultaneously satisfy technical specifications and economic feasibility for industrial applications. The selection of an appropriate catalyst is a significant challenge due to its intricate relationship with the characteristics of the oil feedstock [3,4].
Heterogeneous catalysts demonstrate superior technical advantages over homogeneous catalysts, including minimized corrosion risks, recyclability, and enhanced activity, rendering them ideal for biodiesel synthesis from acidified feedstocks [3]. Keggin-type 12-tungstophosphoric acid (HPW) emerges as a prominent heteropolyacid catalyst due to its structural integrity, synthetic accessibility, environmental compatibility, and strong acidity, delivering consistent catalytic efficiency across reaction systems. Nevertheless, practical implementation remains constrained by intrinsic limitations in surface area and recovery feasibility [5,6].
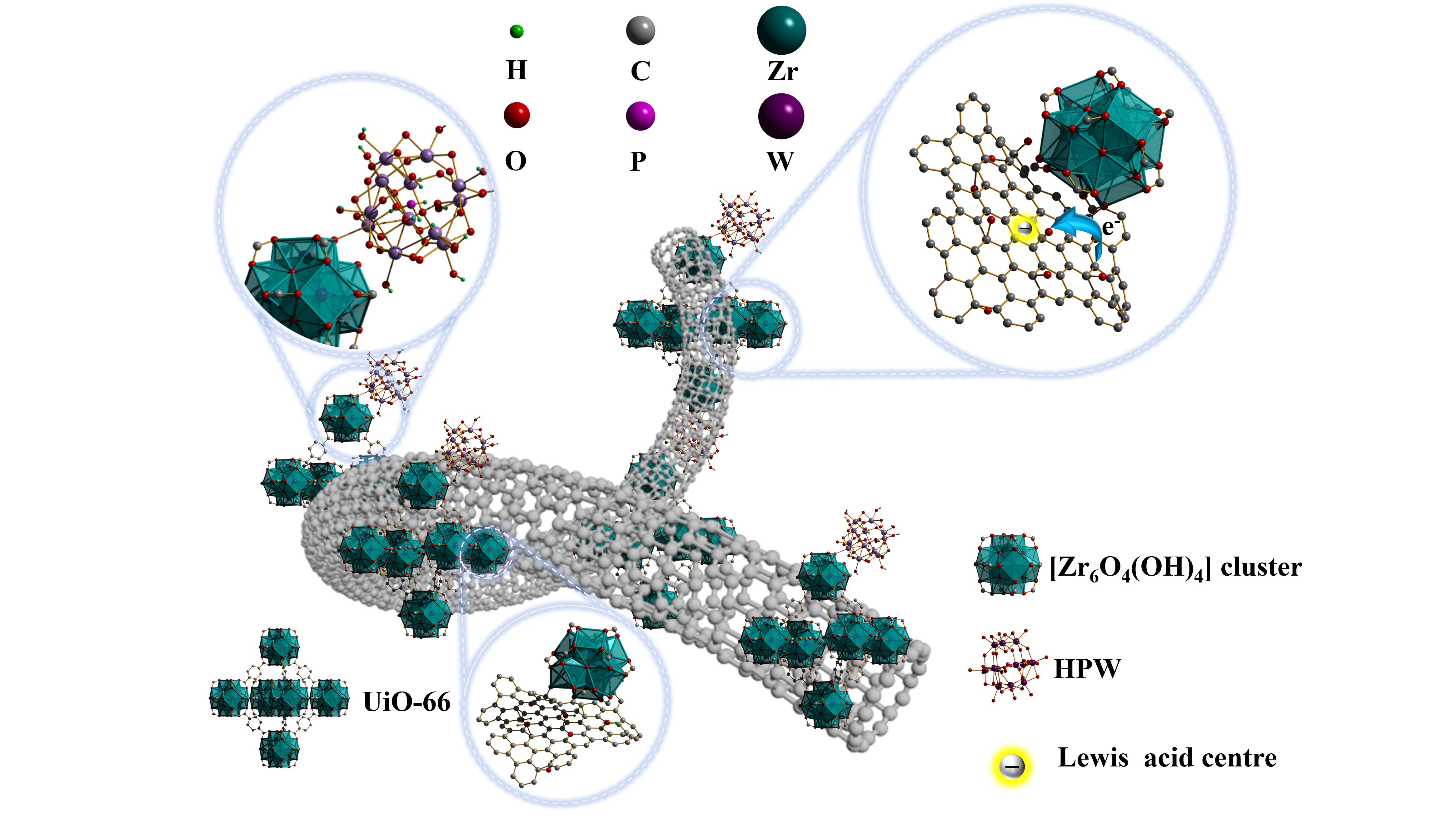
To address HPW loading limitations on multi-walled carbon nanotubes (MWCNTs) while enhancing Lewis acid site density, the research group employs UiO-66 as a bridging agent to bind HPW to MWCNTs. The design rationale originates from the unique electron configuration of MWCNTs. Specifically, MWCNTs are composed of multiple concentric graphite layers with an abundant π-electron system on their surfaces. These π electrons form a dense cloud of active electrons on the surface of MWCNTs, resulting in significantly uneven charge distribution. In particular, the direction perpendicular to the π plane exhibits a negative quadrupole moment (Qzz < 0) due to the dense distribution of the electron cloud. This unique electron cloud structure not only gives MWCNTs excellent electrical conductivity and mechanical strength, but also gives them unique charge interaction capabilities [7]. UiO-66 is a porous metal-organic frame material (MOFs), which is composed of [Zr6O4(OH)4] cluster and organic ligand p-Phthalic acid (PTA) through coordination bonds to form a three-dimensional network structure. The structure contains [Zr6O4(OH)4]12+ secondary structural units, which may interact with the π-electron system on the surface of MWCNTs by van der Waals forces, thereby enhancing the interaction between the two [8]. The unsaturated coordination site of UiO-66 can accept the electron pairs provided by HPW and form stable coordination bonds. HPW can be loaded inside or on the surface of UiO-66 to form a composite material. This composite combines the catalytic activity of HPW with the high specific surface area and pore structure of UiO-66, which improves the catalytic efficiency. Based on the above analysis, a Brönsted-Lewis dual acid catalyst, PW/UiO/CNTs-OH, was synthesized from using HPW, UiO-66, and hydroxylated multi-walled carbon nanotubes (MWCNTs-OH) as raw materials via a one-step hydrothermal method.
2. Materials and Methods
2.1. Materials
12-tungstophosphoric acid (99 %, analytical pure), multi-walled carbon nanotubes (> 95 %, analytical pure), zirconium chloride (98 %, analytical pure) were purchased from Sigma-Aldrich China. Nitric acid (99 %, analytical pure), Glacial acetic acid (99 %, analytical pure), N, N-dimethylformamide (99 %, analytical pure) was purchased from Xirong chemical engineering Co., Ltd. China. Oleic acid (99.8 %, analytical pure) and p-phthalic acid (99 %, analytical pure) were purchased from Shanghai Macklin Biochemical Co., Ltd. China. Methanol (99 %, analytical pure) was purchased from Sinopharm chemical reagent Co., Ltd. China.
2.2. Equipment
Analytical balance (FA1004, Shanghai Lichen Bangxi Instrument Technology Co., Ltd. China); ultrasonic cleaner (KQ-500DE, Kunshan Ultrasonic Instrument Co., Ltd. China); magnetic hotplate stirrer (HWCL-3, Zhengzhou Great Wall Technology and Trade Co., Ltd, China); electro thermal constant-temperature dry box (DHG-9036A, Shanghai Jinghong Experimental Equipment Co., Ltd. China); vacuum oven (DZF-6020, Shanghai YiHeng Scientific Equipment Co., Ltd. China); centrifuge (TGL-16C, Shanghai Anting Science Instrument Factory, China); water-circulating vacuum pump (SHZ-D(III), Shanghai Lichen Bangxi Instrument Technology Co., Ltd. China); gas chromatograph (7820A, Agilent Technology Co., Ltd, America).
2.3. PW/UiO/CNTs-OH Catalyst Preparation
The preparation of MWCNTs-OH catalyst was as follows: Initially,1 g of MWCNTs was dispersed in 60 mL of concentrated HNO3 under mechanical stirring (700 rpm, 1 hour), followed by ultrasonication (30 min). The mixture was filtered, washed with deionized water until the filtrate reached neutrality. Finally, the resulting black powder was dried in a vacuum oven at 60°C for 12 hours to obtain hydroxylated multi-walled carbon nanotubes (MWCNTs-OH).
The preparation of UiO-66 catalyst was as follows: Initially, 0.50 g of ZrCl4 was dissolved in 64 mL of N, N-dimethylformamide (DMF) under continuous stirring (30 min). After adding 8 mL of CH3COOH, the solution was sonicated (30 min). The mixture was then transferred to a Teflon-lined autoclave and heated at 130°C for 4 hours. Subsequently, 0.36 g of PTA (ensuring a molar ratio of ZrCl4: PTA = 1:1) was introduced, and the mixture underwent a second hydrothermal synthesis at 100°C for 36 hours. Finally, the product was collected by centrifugation, washed sequentially with DMF and methanol (three times each), and dried in a vacuum oven at 80°C for 12 hours to obtain UiO-66.
The preparation of PW/UiO-66 catalyst was as follows: Following the initial steps identical to those used for UiO-66 synthesis (dissolution of 0.50 g ZrCl4 in 64 mL DMF, addition of 8 mL CH3COOH, and hydrothermal treatment at 130°C for 4 hours), 0.40 g of HPW and 0.30 g of PTA (ensuring a ZrCl4:PTA molar ratio of 1.2:1) were added after cooling. The mixture was then subjected to a hydrothermal reaction at 100°C for 36 hours. Finally, the product was collected by centrifugation, washed sequentially with DMF and methanol (three times each), and dried in a vacuum oven at 80°C for 12 hours to yield PW/UiO.
The preparation of PW/UiO/CNTs-OH catalyst was as follows: The procedure followed the protocol for PW/UiO synthesis with the following modifications: After the initial hydrothermal step (130°C for 4 hours), 0.01 g of MWCNTs-OH was dispersed into the cooled mixture via ultrasonication for 30 minutes. Subsequently, 0.40 g of HPW and 0.30 g of PTA (ensuring a ZrCl4:PTA molar ratio of 1.2:1) were added to the mixture. The final hydrothermal reaction was conducted at 100°C for 36 hours. The product was then collected by centrifugation, washed sequentially with DMF and methanol (three times each), and dried at 80°C for 12 hours, yielding PW/UiO/CNTs-OH.
2.4. Characterization of the Catalysts
The chemical bonding state in the catalyst was determined by fourier transform infrared spectroscopy (FT-IR) (Nicolet iS50, Thermo Fisher, American) with a scanning range of 4000-400 cm-1. The samples were prepared by KBr pellet method at room temperature. The crystalline phases of the catalyst were analyzed by X-ray diffraction (XRD) (SmartLab SE, Nippon Science) with Cu Kα radiation at 3 kW at a scanning range of 5-85°. The elemental composition, valence state and content of catalysts were determined by X-ray photoelectron spectroscopy (XPS) (Thermo Scientific K-Alpha, Thermo Fisher, American) under the vacuum condition of P < 10-6 Pa. A monochromatic Al Kα X-ray source (hv=1486.6 eV) was used with a power of 100 W. The content of W in the catalyst was analyzed by inductively coupled plasma optical emission spectrometer (ICP-OES) (Thermo ICAP PRO, Thermo Fisher, American). The morphology of catalysts was observed by scanning electron microscope (SEM) (MLA650F, FEI company, American). The affinity of catalyst to deionized water was tested by optical contact angle measuring instrument (JYC, ShangHai FangRui Instrument Ltd, China). The nitrogen adsorption-desorption isotherm of catalysts was measured by N2 adsorption instrument (ASAP2460, Micromeritics Instrument Ltd.) at 77 K to determine the specific surface area, pore volume, and pore size distribution. The content and strength of acidic active sites of catalysts were analyzed by NH3 temperature programmed desorption (NH3-TPD) (AutoChem II 2920, Micromeritics Instrument Ltd., American) with the temperature range of 50-750°C. The Brӧnsted acid sites and Lewis acid sites on the surface of catalysts were measured by pyridine adsorption IR spectra (Py-IR) (Nicolet iS50, Thermo Fisher, American). The electrochemical impedance tests (EIS) were conducted by using an electrochemical workstation (CHI660D, ShangHai HuaChen Ltd, China). The weight loss and stability of the samples at different temperatures were determined by thermal gravimetric analyzer (TG) (Netzsch, STA449F5, Selb, Germany).
2.5. Esterification Process Catalyzed by PW/UiO/CNTs-OH
The esterification process was conducted as follows: The oleic acid and methanol were precisely mixed at a predetermined molar ratio. Subsequently, the homogeneous mixture along with a specified amount of catalyst was transferred into a sealed stainless steel autoclave reactor. The reaction system was maintained under isothermal conditions for a predetermined duration.
Upon completion of the reaction, 1 mL of crude product was collected and subjected to solvent evaporation in a drying oven (60°C, 1 h) to remove residual methanol and aqueous-phase byproducts. The concentrated sample was then diluted to 5 mL with HPLC-grade methanol to ensure homogeneity. The resulting solution was filtered through a 0.45 μm membrane filter to eliminate particulate matter and residual catalyst particles. Finally, the concentration of FAME was determined by gas chromatography and quantified based on the external standard method. The formula for calculating the yield (%) is as follows:
Where, m0 is the initial mass of oleic acid (g), mi is the mass of FAME after reacting a certain time (g), mMO are the mass of methyl oleate.
Upon completion and cooling of the reaction, the resulting mixture separated into two distinct layers. The upper layer consisted of FAME and a minor amount of unreacted oleic acid, while the lower layer was a turbid mixture containing the solid catalyst PW/UiO/CNTs-OH, methanol, and water. To recover the catalyst, PW/UiO/CNTs-OH was isolated from the mixture via suction filtration, washed three times with tert-butanol, subsequently reactivated at 130°C for reuse.
2.6. Gas Chromatographic Conditions
Agilent 7820A gas chromatography with DB-5 column (30 m×0.25 mm×0.25μm) was used to detect the target product. The injection volume was 1μL in a split flow of 20:1. The column oven was initially held at 50°C followed by heating to 180°C at a heating rate of 20°C/min, then increased to 200°C at 2°C/min, and finally heated to 250°C at a rate of 10°C/min and held for 5 min. Nitrogen was used as carrier gas at a rate of 25 mL/min.
2.7. Experimental Design
In this study, the esterification reaction parameters were optimized using central composite design-response surface methodology (CCD-RSM). A three-factor, five-level experimental framework was implemented through Design Expert software (version 10.0.7.0) for both design generation and statistical analysis. The central composite design examined the effects of molar ratio of methanol to oleic acid (X1, mol/mol), catalyst loading (X2, wt%), and reaction temperature (X3, °C) on methyl oleate yield (Y, %) were investigated through CCD, employing a second-order polynomial model (equation (2)). A total of 20 experiments were designed (20=2n+2n+n0, where n=3 factors and n0=6 center points), including 8 factorial points, 6 axial points, and 6 repeated central points. The model's adequacy was assessed via analysis of variance (ANOVA) and significance tests. The levels of independent variables and their experimental ranges are summarized in Table 1.
Where Y is the expected response value (methyl oleate yield) and α0 and αi are the coefficient constant and linear constant, respectively. αii and αij terms are the quadratic coefficients and interaction coefficients respectively. The terms Xi and Xj denote independent parameters [9].
3. Results
3.1. Catalyst Characterization
XRD was performed to characterize the crystalline phases of the synthesized samples. Figure 1 shows the XRD patterns of the different materials and the standard XRD pattern of UiO-66, generated from the original CIF file [10]. The observed diffraction peaks of UiO-66 at 7.36°, 8.51°, 25.74°, and 30.78° were in excellent agreement with the simulated pattern reported in reference [11]. After HPW loading, the crystallinity of PW/UiO was significantly reduced. This was due to the addition of HPW during catalyst preparation, which reduced the pH of the solution, resulting in the easy protonation of terephthalic acid. As a result, the coordination of the [Zr6O4(OH)4] cluster was inhibited, leading to a reduction in crystallinity [8]. Furthermore, the XRD patterns of HPW could not be found in the XRD patterns of the PW/UiO-66 and PW/UiO/CNTs-OH samples, indicating an even distribution of HPW [12].
The morphological features of synthesized materials HPW, MWCNTs-OH, UiO-66, PW/UiO and PW/UiO/CNTs-OH were characterized by SEM (Figure 2(a-e)). At the same time, the hydrophilicity of MWCNTs, MWCNTs-OH, UiO-66, PW/UiO, and PW/UiO/CNTs-OH was determined by optical contact angle measuring instrument (Figure 2(f-j)). The results indicated that the contact angles of the MWCNTs (Figure 2(f)) and MWCNTs-OH (Figure 2(g)) were 140° and 35°, respectively, demonstrating a significant enhancement in the hydrophilicity of MWCNTs-OH, which confirmed successful hydroxylation. This was because the addition of HPW inhibited the coordination of the terephthalic acid ligand with the [Zr6O4(OH)4] cluster, resulting in an irregular shape and easy agglomeration of PW/UiO [8], while enhancing the hydrophilicity of the material (contact angle of 72°). When MWCNTs-OH was incorporated into PW/UiO (Figure 2(e)), it was encapsulated within the composite, further enhancing the material's hydrophilicity (contact angle of 54°). However, compared to MWCNTs-OH alone, the hydrophilicity of PW/UiO/CNTs-OH was slightly diminished, possibly because of interactions between UiO-66 and the hydrophilic groups on MWCNTs-OH [13].
The FT-IR spectra of the MWCNTs, HPW, UiO-66, PW/UiO, and PW/UiO/CNTs-OH are shown in Figure 3. For UiO-66, the broad bands spanning 2500-3700 cm-1 corresponded to O-H stretching vibrations from Zr-OH groups [14], while the absorption peaks at 1584 cm-1 and 1391 cm-1 were attributed to the asymmetric and symmetric O=C-O stretching modes of the carboxylic acid groups in PTA. The band at 1660 cm-1 aroused from residual DMF solvent molecules within the framework, as evidenced by its characteristic C=O stretching vibration [10]. The absorption band observed at 1505 cm-1 was due to the C=C stretching vibration within the benzene ring, whereas the band at 1098 cm-1 corresponded to the Zr-O vibrational mode. The characteristic peaks at 822 cm-1, 743 cm-1, and 550 cm-1 were respectively assigned to C-H deformation, C=C stretching, and Zr-(OC) asymmetric stretching vibrations [12,15]. After HPW loading, the characteristic bands of the Keggin-structured polyoxometalate anion at 1072 cm-1 (P=O), 955 cm-1 (W=O), 882 cm-1 (W-Oc-W, where Oc represents angularly shared oxygen atoms), and 715 cm-1 (W-Oe-W, where Oe represents edge-shared oxygen atoms) were observed, indicating that HPW was successfully loaded onto UiO-66. Notably, the emergence of a new absorption band at 648 cm-1, attributed to the Zr-O-W stretching vibration, accompanied by blue shifts of the Zr-O band at 1098 cm-1 and the W-Oc-W band at 882 cm-1 confirms the coordination bonding between the zirconium sites in UiO-66 and the [PW12O40]3- heteropoly anion. These spectral modifications demonstrate the electron density redistribution resulting from Zr to W charge transfer interactions [16,17]. The carrier, MWCNTs-OH, was further introduced, and it was found that these two bands experienced a further blue shift. This may be due to the dense and reactive electron clouds on the surface of the MWCNTs, which created a pronounced uneven charge distribution within the π-plane, characterized by a negative quadrupole moment perpendicular to the plane (Qzz < 0). This phenomenon inherently enabled the MWCNTs to attract and stabilize [Zr6O4(OH)4]12+ [7]. Consequently, the Zr4+ ions exhibited a pronounced strong interaction with the π electrons of the MWCNTs, leading to electron cloud displacement towards Zr. This phenomenon resulted in the weakening of the Zr-O bond and a subsequent reduction in the vibration frequency.
The valence states and chemical bonding configurations of the elements in the MWCNTs-OH, Zr-CNTs-OH, UiO/CNTs-OH, and PW/UiO/CNTs-OH catalysts were investigated using XPS (Figure 4(a-d)). The C 1s spectra of the samples are shown in Figure 4(a). The C 1s spectrum of MWCNTs-OH primarily comprised three distinct peaks. Specifically, the predominant peak at 284.80 eV corresponded to C=C and C-C bonds, while those at 286.90 eV and 291.13 eV corresponded to C-O and π-π* bonds, respectively. In Zr-functionalized derivatives (Zr-CNTs-OH, UiO/CNTs-OH, PW/UiO/CNTs-OH), the C-O binding energy exhibited a 0.40 eV downshift from 286.90 eV to 286.50 eV relative to MWCNTs-OH. Correspondingly, the O 1s spectrum of Zr-CNTs-OH (Figure 4b) displayed a 0.46 eV C-O energy reduction from 532.66 eV to 532.20 eV, arising from Zr covalent bonding at the MWCNTs-OH defect sites. The formation of C-O-Zr interactions weakened the C-O bond strength, thereby leading to a reduction in the binding energy [13,15]. The O 1s spectrum of UiO/CNTs-OH exhibited two distinct peaks. Specifically, the peak at a binding energy of 531.91 eV corresponded to Zr-Oa-C (where Oa represents oxygen in carboxylic acid groups), while the peak at 530.16 eV was attributed to Zr-Ob (where Ob denotes lattice oxygen) [18]. HPW incorporation induced a 0.10 eV binding energy decrease in Zr-Oa-C (531.91 → 531.81 eV) and a 0.18 eV increase in Zr-Ob (530.16 → 530.34 eV), demonstrating an interaction between HPW and UiO-66 [19]. The Zr 3d spectra of samples are presented in Figure 4(c). In conjunction with the C 1s spectral peak analysis, it confirmed that Zr clusters had been successfully loaded onto MWCNTs-OH. Notably, after the introduction of HPW, the Zr 3d3/2 peak (185.09 eV) and the Zr 3d5/2 peak (182.71 eV) in PW/UiO/CNTs-OH shifted to higher binding energies, reaching 186.30 eV and 183.91 eV, respectively, compared to UiO/CNTs-OH. The W 4f spectrum of PW/UiO/CNTs-OH, shown in Figure 4(d), revealed 0.91 eV (W 4f7/2: 35.8 → 36.71 eV) and 0.88 eV (W 4f5/2: 37.9 → 38.78 eV) increases relative to pristine HPW [20]. Additionally, the peak observed at 42.18 eV corresponded to Zr-O-W [21], confirmed oxygen-bridged coordination between Zr and [PW₁₂O₄₀]³⁻ heteropolyanions, consistent with FTIR analysis.
The W content in PW/CNTs, PW/CNTs-OH, PW/UiO, and PW/UiO/CNTs-OH was systematically analyzed using ICP-OES (Table 2). It was found that the W contents were determined to be 1.49 wt% and 1.67 wt% for PW/CNTs and PW/CNTs-OH, respectively, indicating that MWCNTs exhibited limited HPW loading capacity regardless of their hydroxylation status. In contrast, significantly higher W contents of 4.39 wt% and 14.87 wt% were observed in PW/UiO and PW/UiO/CNTs-OH, respectively. The remarkable increase in W content after MWCNTs-OH incorporation was attributed to the π-π stacking interactions between MWCNTs and PTA. Furthermore, it was demonstrated that MWCNTs-OH could effectively anchor [Zr6O4(OH)4] clusters through C-O-Zr bonding, which provided additional nucleation sites for UiO-66 synthesis and consequently enhanced the encapsulation efficiency of HPW within UiO-66 [22].
N2 adsorption-desorption tests were conducted to evaluate the textural properties of MWCNTs-OH, UiO-66, PW/UiO, and PW/UiO/CNTs-OH catalysts (Figure 5a-d). The adsorption and desorption isotherms of UiO-66 (Figure 5(b)) exhibited type-I isotherm, characteristic of microporous materials, whereas both PW/UiO (Figure 5c) and PW/UiO/CNTs-OH (Figure 5d) displayed type-IV isotherms with hysteresis loops (P/P0 = 0.45-1.0), indicating mesoporous structures. Upon loading HPW, the specific surface area decreased from 997.531 m2/g to 700.046 m2/g (Table 3), while the average pore size increased from 2.197 nm to 3.173 nm (Table 3). Subsequent MWCNTs-OH incorporation enhanced the surface area to 857.147 m2/g (Table 3), improving the reactant-catalyst contact efficiency.
The acidic properties of UiO-66, PW/UiO, and PW/UiO/CNTs-OH were characterized by NH3-TPD and Py-IR. As shown in Table 4 and Figure 6a, the acidic distribution and total acidity were quantified. Three distinct ammonia desorption peaks were observed for UiO-66 in the temperature ranges of 25-120°C, 120-310°C, and 310-450°C. The absorption peak at 25-120°C corresponded to the weak acid sites, attributed to the weak Brönsted acidic sites originating from the framework μ3-OH [18]. The 120-310°C desorption peak was assigned to Lewis acid sites created by unsaturated Zr sites formed owing to the lack of organic linkers in UiO-66. The medium-strong acid absorption peak at 310-450°C was derived from the Brönsted acid sites associated with Zr-OH [23,24,25]. Acid characterization revealed significant modifications in PW/UiO compared to UiO-66. The weak acid content decreased from 4.57 to 3.64 mmol/g, while the medium-strength acid content increased from 1.23 to 2.58 mmol/g. Concurrently, the Lewis acid density rose from 34.72 to 44.54 μmol/g (Figure 6(b)). This acid modification was likely attributed to the interaction between the heteropolyacid anion [PW12O40]3- and Zr, which resulted in a decrease in the number of μ3-OH sites and an increase in unsaturated Zr sites, thereby enhancing the Lewis acidity. Concurrently, the inherent Brönsted acidity of HPW was found to augment the corresponding acid sites in PW/UiO [26]. Significant acid enhancement was observed in PW/UiO/CNTs-OH following MWCNTs-OH incorporation, with weak acid content increasing from 3.64 to 4.36 mmol/g and medium-strength acidity rising from 2.58 to 3.05 mmol/g. The Brönsted acid density increased from 3.65 to 9.98 μmol/g, while the Lewis acidity nearly doubled to 83.63 μmol/g (Figure 6(b)). This improvement was mediated by the strong electron-withdrawing effect of the [Zr6O4(OH)4] cluster on the highly delocalized π electrons present in MWCNTs-OH [7], which intensified the Lewis acidity. Additionally, the carbon atoms in MWCNTs-OH could coordinate with [Zr6O4(OH)4]12+ through oxygen, and a π-π stacking interaction existed between MWCNTs-OH and PTA. These interactions provided additional nucleation sites for the synthesis of UiO-66 [22], enabling greater HPW encapsulation and consequent Brönsted acid enhancement.
Electrochemical characterization was conducted to assess charge transfer behavior in UiO-66, PW/UiO, and PW/UiO/CNTs-OH catalysts. As shown in Figure 6(c), the electron transfer efficiency followed the sequence: UiO-66 > PW/UiO > PW/UiO/CNTs-OH. Notably, PW/UiO/CNTs-OH exhibited the smallest EIS arc diameter, indicating superior electron migration efficiency, which facilitated the reaction process.
The thermal stability of MWCNTs-OH, UiO-66, PW/UiO, and PW/UiO/CNTs-OH was evaluated by TG. As shown in Figure 6(d), all the samples exhibited three distinct steps in the weight loss stages. For UiO-66, three weight-loss stages were observed in the temperature ranges of 26-100°C, 100-300°C, and 485-610°C, which were centered at 55, 222, and 557°C, respectively. The initial stage (26–100°C) was attributed to the evaporation of the physically adsorbed H2O and methanol. This was followed by a weight-loss stage (100–300°C) resulting from DMF solvent removal from the UiO-66 pores and decomposition of residual PTA [18]. Structural collapse and thermal degradation of the framework to ZrO2 were observed in the final stage (485–610°C) [11]. In addition, no further weight loss was detected above 610°C. The PW/UiO-66 sample exhibited identical first two stages of decomposition to UiO-66, with its third stage of mass loss (400-610°C) attributed to the structural collapse of the framework combined with HPW decomposition into WO3 and P2O5 [27]. Collectively, the thermal stability analysis confirmed that all materials maintained structural integrity below 400°C.
The formation mechanism of the Lewis acid sites in PW/UiO/CNTs-OH and the bridging function of UiO-66 are schematically illustrated in Figure 7. Specifically, the [Zr6O4(OH)4] cluster within UiO-66 coordinates with tungsten atoms in the [PW₁₂O₄₀]⁻ polyoxometalate anion via oxygen linkages, anchoring HPW. Simultaneously, it formed oxygen-mediated coordination bonds with MWCNTs-OH. This dual coordination mechanism effectively bridges HPW and MWCNTs-OH. Simultaneously, the π-electrons of MWCNTs exhibited a strong electron-withdrawing effect on the [Zr6O4(OH)4] cluster, causing electrons to migrate to the surface of MWCNTs and delocalize within their aromatic structures. This enhanced the charge imbalance in the PW/UiO/CNTs-OH catalyst system, leading to an excess positive charge. This enhances the charge imbalance in the PW/UiO/CNTs-OH catalyst system, leading to an excess positive charge. Ultimately, this results in the formation of Lewis acid sites in the region where the MWCNTs adsorb the [Zr6O4(OH)4] cluster, significantly improving the catalytic activity.
3.2. Catalytic Activity
The catalytic performance of UiO-66, PW/UiO, and PW/UiO/CNTs-OH in esterification reactions was evaluated under standardized conditions (a methanol to oleic acid molar ratio of 14:1, a catalyst loading of 6 wt%, and a reaction temperature of 70°C), as illustrated in Figure 8. Notably, PW/UiO/CNTs-OH exhibited superior catalytic activity, with a methyl oleate yield of 92.0%. This enhanced performance could be attributed to its higher concentration of acidic sites and enhanced improved electron migration efficiency compared to UiO-66 and PW/UiO.
A central composite design was implemented with three independent variables: methanol-to-oleic acid molar ratio (X1, mol/mol), catalyst loading (X2, wt%), and reaction temperature (X3, °C), while the yield of FAME (Y, %) served as the response variable. The experimental matrix containing 20 experimental runs and corresponding results are systematically presented in Table 6, with the ANOVA analysis for the RSM detailed in Table 7. Through statistical evaluation, the quadratic regression equation was derived as follows:
This study evaluated the model fit and the prediction confidence of each item through the p-value, F-value, and R2 metrics. A correlation coefficient R2 0.9975 was obtained, demonstrating the model's capability to reliably predict methyl oleate yields. Moreover, close agreement between the Pred R2 (0.9808) and the Adj R2 (0.9953), with a difference of < 0.2, confirmed the model's effectiveness in representing actual factor-response relationships. This minimal discrepancy further substantiated the formula's goodness of fit. Experimental repeatability was verified by a coefficient of variation (C.V. = 1.60%) below the 2% acceptability threshold.
The effect of catalyst loading, methanol to oleic acid molar ratio and reaction temperature on biodiesel yield are presented in Figure 9(a-c). The interaction between catalyst loading and the molar ratio of methanol to oleic acid under fixed reaction duration at the central point (70°C) was displayed in Figure 9(a). A significant enhancement in methyl oleate yield was observed when catalyst loading increased from 2.6 wt% to 6 wt% concurrently with the molar ratio elevation from 10.6:1 to 14:1. However, further increases to 10 wt% catalyst and 17.3:1 molar ratio caused marginal yield reduction, attributable to the reversible nature of the esterification reaction. Heightening reactant ratios thermodynamically favor forward conversion while optimal catalyst dosage maximizes active site utilization. In the esterification system, methanol concentration plays a dominant role. In the esterification system, methanol concentration plays a dominant role. The methyl oleate yield increases with methanol addition, but when methanol concentration exceeds critical levels, adsorbed methanol accumulates on the catalyst surface. This accumulation reduces the oleic acid-catalyst contact area and may ultimately induce catalyst deactivation through active site blockage [28]. Furthermore, the maximum methyl oleate yield of 92.0% was achieved at 6 wt% catalyst loading, beyond which no significant enhancement was observed despite increased catalyst loading [29]. A temperature-dependent enhancement was demonstrated in Figure 9(c), where methyl oleate yield showed progressive improvement with elevated temperatures due to the endothermic nature of esterification that thermodynamically favors forward reaction kinetics. The parameter significance ranking derived from Figure 9 demonstrated X2 > X1 > X3. The model-predicted optimal parameters (14.3:1 molar ratio of methanol to oleic acid, 6.54 wt% catalyst loading, 68.8°C reaction temperature) were determined to yield 91.9% methyl oleate through balanced activation energy and mass transfer optimization.
The reusability of PW/UiO/CNTs-OH was evaluated under optimal reaction conditions, after which the catalyst was centrifugally recovered and triple-washed with tert-butanol to eliminate surface-adsorbed methyl oleate and residual oleic acid, followed by vacuum activation at 130°C for 12 hours. The model-predicted methyl oleate yield of 91.9% showed close alignment with the experimental value (92.9%), with a deviation of only 1.0% demonstrating model accuracy. As evidenced in Figure 10, a yield reduction to 82.3% was observed after the fourth reuse cycle, attributed to potential pore channel blockage that compromised active site accessibility.
4. Conclusions
In this study, a Brönsted-Lewis solid acid catalyst PW/UiO/CNTs-OH was successfully synthesized through UiO-66-mediated bridging of HPW and MWCNTs. Under optimal conditions, where the molar ratio of methanol to oleic acid was 14.27:1, the catalyst loading was 6.54 wt%, the reaction temperature was 68.8°C, and the reaction time was 90 minutes, the yield of methyl oleate reached 92.9%. Furthermore, after four cycles, the catalyst retained a methyl oleate yield of 82.3%, demonstrating operational stability. The enhanced performance of PW/UiO/CNTs-OH originated from electron-withdrawing interactions between the [Zr6O4(OH)4] clusters in UiO-66 and the π-electrons of MWCNTs, which promoted the formation of numerous strong Lewis acid sites. This work introduced an MOF-based chemical bridging strategy that establishes a groundbreaking paradigm for heterogeneous catalyst design, thereby substantially expanding viable industrial applications.
Author Contributions
Conceptualization, Xuyao Xing. and Qing Shu.; Methodology, Xuyao Xing.; Software, Li Zhang.; Validation, Qiong Wu., Xuyao Xing.; Formal Analysis, Qiong Wu.; Investigation, Li Zhang.; Resources, Qing Shu.; Data Curation, Qiong Wu.; Writing – Original Draft Preparation, Xuyao Xing.; Writing – Review & Editing, Qing Shu.; Visualization, Qiong Wu.; Supervision, Qing Shu.; Project Administration, Li Zhang.; Funding Acquisition, Qing Shu.
Funding
The National Natural Science Foundation of China (21766009).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21766009).
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abdullah, S.H.Y.S.; Hanapi, N.H.M.; Azid, A.; Umar, R.; Juahir, H.; Khatoon, H.; Endut, A. A Review of Biomass-Derived Heterogeneous Catalyst for a Sustainable Biodiesel Production. Renewable and Sustainable Energy Reviews 2017, 70, 1040–1051. [Google Scholar] [CrossRef]
- Bergmann, J.C.; Tupinambá, D.D.; Costa, O.Y.A.; Almeida, J.R.M.; Barreto, C.C.; Quirino, B.F. Biodiesel Production in Brazil and Alternative Biomass Feedstocks. Renewable and Sustainable Energy Reviews 2013, 21, 411–420. [Google Scholar] [CrossRef]
- Zabeti, M.; Wan Daud, W.M.A.; Aroua, M.K. Activity of Solid Catalysts for Biodiesel Production: a Review. Fuel Processing Technology 2009, 90, 770–777. [Google Scholar] [CrossRef]
- Nath, B.; Das, B.; Kalita, P.; Basumatary, S. Waste to Value Addition: Utilization of Waste Brassica nigra Plant Derived Novel Green Heterogeneous Base Catalyst for Effective Synthesis of Biodiesel. Journal of Cleaner Production 2019, 239, 118112. [Google Scholar] [CrossRef]
- Frattini, L.; Isaacs, M.A.; Parlett, C.M.A.; Wilson, K.; Kyriakou, G.; Lee, A.F. Support Enhanced α-pinene Isomerization over HPW/SBA-15. Applied Catalysis B: Environmental 2017, 200, 10–18. [Google Scholar] [CrossRef]
- You, X.; Yu, L.-l.; Xiao, F.-f.; Wu, S.-c.; Yang, C.; Cheng, J.-h. Synthesis of Phosphotungstic Acid-Supported Bimodal Mesoporous Silica-Based Catalyst for Defluorination of Aqueous Perfluorooctanoic Acid under Vacuum UV Irradiation. Chemical Engineering Journal 2018, 335, 812–821. [Google Scholar] [CrossRef]
- Bornhof, A.-B.; Bauzá, A.; Aster, A.; Pupier, M.; Frontera, A.; Vauthey, E.; Sakai, N.; Matile, S. Synergistic Anion–(π)n–π Catalysis on π-Stacked Foldamers. Journal of the American Chemical Society 2018, 140, 4884–4892. [Google Scholar] [CrossRef]
- Ullah, L.; Zhao, G.; Hedin, N.; Ding, X.; Zhang, S.; Yao, X.; Nie, Y.; Zhang, Y. Highly Efficient Adsorption of Benzothiophene from Model Fuel on a Metal-Organic Framework Modified with Dodeca-Tungstophosphoric Acid. Chemical Engineering Journal 2019, 362, 30–40. [Google Scholar] [CrossRef]
- Arrais Gonçalves, M.; Karine Lourenço Mares, E.; Roberto Zamian, J.; Narciso da Rocha Filho, G.; Rafael Vieira da Conceição, L. Statistical Optimization of Biodiesel Production from Waste Cooking Oil Using Magnetic Acid Heterogeneous Catalyst MoO3/SrFe2O4. Fuel 2021, 304. [Google Scholar] [CrossRef]
- Valenzano, L.; Civalleri, B.; Chavan, S.; Bordiga, S.; Nilsen, M.H.; Jakobsen, S.; Lillerud, K.P.; Lamberti, C. Disclosing the Complex Structure of UiO-66 Metal Organic Framework: a Synergic Combination of Experiment and Theory. Chemistry of Materials 2011, 23, 1700–1718. [Google Scholar] [CrossRef]
- Man, Z.; Meng, Y.; Lin, X.; Dai, X.; Wang, L.; Liu, D. Assembling UiO-66@TiO2 Nanocomposites for Efficient Photocatalytic Degradation of Dimethyl Sulfide. Chemical Engineering Journal 2022, 431, 133952. [Google Scholar] [CrossRef]
- Ma, Y.; Li, A.; Wang, C.; Ge, X. Preparation of HPW@UiO-66 Catalyst with Defects and its Application in Oxidative Desulfurization. Chemical Engineering Journal 2021, 404, 127062. [Google Scholar] [CrossRef]
- Zeng, Z.; Sorescu, D.C.; White, D.L.; Hwang, S.I.; Shao, W.; He, X.; Schulte, Z.M.; Rosi, N.L.; Star, A. Heterogeneous Growth of UiO-66-NH2 on Oxidized Single-Walled Carbon Nanotubes to Form “Beads-on-a-String” Composites. ACS Applied Materials & Interfaces 2021, 13, 15482–15489. [Google Scholar] [CrossRef]
- Alcañiz-Monge, J.; Bakkali, B.E.; Trautwein, G.; Reinoso, S. Zirconia-supported Tungstophosphoric Heteropolyacid as Heterogeneous Acid Catalyst for Biodiesel Production. Applied Catalysis B: Environmental 2018, 224, 194–203. [Google Scholar] [CrossRef]
- Chen, C.; Chen, D.; Xie, S.; Quan, H.; Luo, X.; Guo, L. Adsorption Behaviors of Organic Micropollutants on Zirconium Metal–Organic Framework UiO-66: Analysis of Surface Interactions. ACS Applied Materials & Interfaces 2017, 9, 41043–41054. [Google Scholar] [CrossRef]
- Rao, K.N.; Sridhar, A.; Lee, A.F.; Tavener, S.J.; Young, N.A.; Wilson, K. Zirconium Phosphate Supported Tungsten Oxide Solid Acid Catalysts for the Esterification of Palmitic Acid. Green Chemistry 2006, 8. [Google Scholar] [CrossRef]
- Yan, L.; Duan, T.; Huang, T.; Zhao, B.; Fan, Y. Phosphotungstic Acid Immobilized on Mixed-Ligand-Directed UiO-66 for the Esterification of 1-butene with Acetic Acid to Produce High-Octane Gasoline. Fuel 2019, 245, 226–232. [Google Scholar] [CrossRef]
- Kar, A.K.; Sarkar, R.; Manal, A.K.; Kumar, R.; Chakraborty, S.; Ahuja, R.; Srivastava, R. Unveiling and Understanding the Remarkable Enhancement in the Catalytic Activity by the Defect Creation in UiO-66 during the Catalytic Transfer Hydrodeoxygenation of Vanillin with Isopropanol. Applied Catalysis B: Environmental 2023, 325, 122385. [Google Scholar] [CrossRef]
- Ma, T.; Liu, D.; Liu, Z.; Xu, J.; Dong, Y.; Chen, G.; Yun, Z. 12-Tungstophosphoric Acid-Encapsulated Metal-Mrganic Framework UiO-66: A Promising Catalyst for the Esterification of Acetic Acid with N-butanol. Journal of the Taiwan Institute of Chemical Engineers 2022, 133. [Google Scholar] [CrossRef]
- Liao, X.; Huang, Y.; Zhou, Y.; Liu, H.; Cai, Y.; Lu, S.; Yao, Y. Homogeneously Dispersed HPW/Graphene for High Efficient Catalytic Oxidative Desulfurization Prepared by Electrochemical Deposition. Applied Surface Science 2019, 484, 917–924. [Google Scholar] [CrossRef]
- Li, M.; Wang, T.; Liu, X.-L.; Bao, Z.-L.; Qian, P.-F.; Liu, K.; Shi, Y.; Ming, X.; Geng, H.-Z. Highly Stable Phosphotungstic Acid/Au Dual Doped Carbon Nanotube Transparent Conductive Films for Transparent Flexible Heaters. Carbon 2023, 207, 219–229. [Google Scholar] [CrossRef]
- Ellis, J.E.; Zeng, Z.; Hwang, S.I.; Li, S.; Luo, T.-Y.; Burkert, S.C.; White, David L. ; Rosi, N.L.; Gassensmith, J.J.; Star, A. Growth of ZIF-8 on Molecularly Ordered 2-Methylimidazole/Single-Walled Carbon Nanotubes to Form Highly Porous, Electrically Conductive Composites. Chemical Science 2019, 10, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New Zirconium Inorganic Building Brick Forming Metal Organic Frameworks with Exceptional Stability. Journal of the American Chemical Society 2008, 130, 13850–13851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zheng, S.; Xiao, Q.; Zhong, Y.; Zhu, W.; Lin, A.; Samy El-Shall, M. Synergetic Catalysis of Palladium Nanoparticles Encaged within Amine-Functionalized UiO-66 in the Hydrodeoxygenation of Vanillin in Water. Green Chemistry 2016, 18, 2900–2908. [Google Scholar] [CrossRef]
- Liang, J.; Chen, R.-P.; Wang, X.-Y.; Liu, T.-T.; Wang, X.-S.; Huang, Y.-B.; Cao, R. Postsynthetic Ionization of an Imidazole-Containing Metal–Organic Framework for the Cycloaddition of Carbon Dioxide and Epoxides. Chemical Science 2017, 8, 1570–1575. [Google Scholar] [CrossRef]
- Freitas, E.F.; Araújo, Á.A.L.; Paiva, M.F.; Dias, S.C.L.; Dias, J.A. Comparative Acidity of BEA and Y Zeolite Composites with 12-tungstophosphoric and 12-tungstosilicic Acids. Molecular Catalysis 2018, 458, 152–160. [Google Scholar] [CrossRef]
- Yang, X.-L.; Qiao, L.-M.; Dai, W.-L. Phosphotungstic Acid Encapsulated in Metal-Organic Framework UiO-66: An Effective Catalyst for the Selective Oxidation of Cyclopentene to Glutaraldehyde. Microporous and Mesoporous Materials 2015, 211, 73–81. [Google Scholar] [CrossRef]
- Niu, S.; Ning, Y.; Lu, C.; Han, K.; Yu, H.; Zhou, Y. Esterification of Oleic Acid to Produce Biodiesel Catalyzed by Sulfonated Activated Carbon from Bamboo. Energy Conversion and Management 2018, 163, 59–65. [Google Scholar] [CrossRef]
- Jiang, Y.; Lu, J.; Sun, K.; Ma, L.; Ding, J. Esterification of Oleic Acid with Ethanol Catalyzed by Sulfonated Cation Exchange Resin: Experimental and Kinetic Studies. Energy Conversion and Management 2013, 76, 980–985. [Google Scholar] [CrossRef]
Figure 1.
XRD pattern of HPW, MWCNTs-OH, PW/UiO/CNTs-OH, PW/UiO, UiO-66 and simulate UiO-66

Figure 2.
SEM images of samples: (a)HPW, (b)MWCNTs, (c)UiO-66, (d)PW/UiO, (e)PW/UiO/CNTs-OH, Water contact angle measurements for (f)MWCNTs, (g)MWCNTs-OH, (h)UiO-66, (i)PW/UiO, (j)PW/UiO/CNTs-OH.
Figure 2.
SEM images of samples: (a)HPW, (b)MWCNTs, (c)UiO-66, (d)PW/UiO, (e)PW/UiO/CNTs-OH, Water contact angle measurements for (f)MWCNTs, (g)MWCNTs-OH, (h)UiO-66, (i)PW/UiO, (j)PW/UiO/CNTs-OH.

Figure 3.
FT-IR spectra (a) and detail (b) of PW/UiO/CNTs-OH, PW/UiO, UiO-66, HPW and MWCNTs-OH.
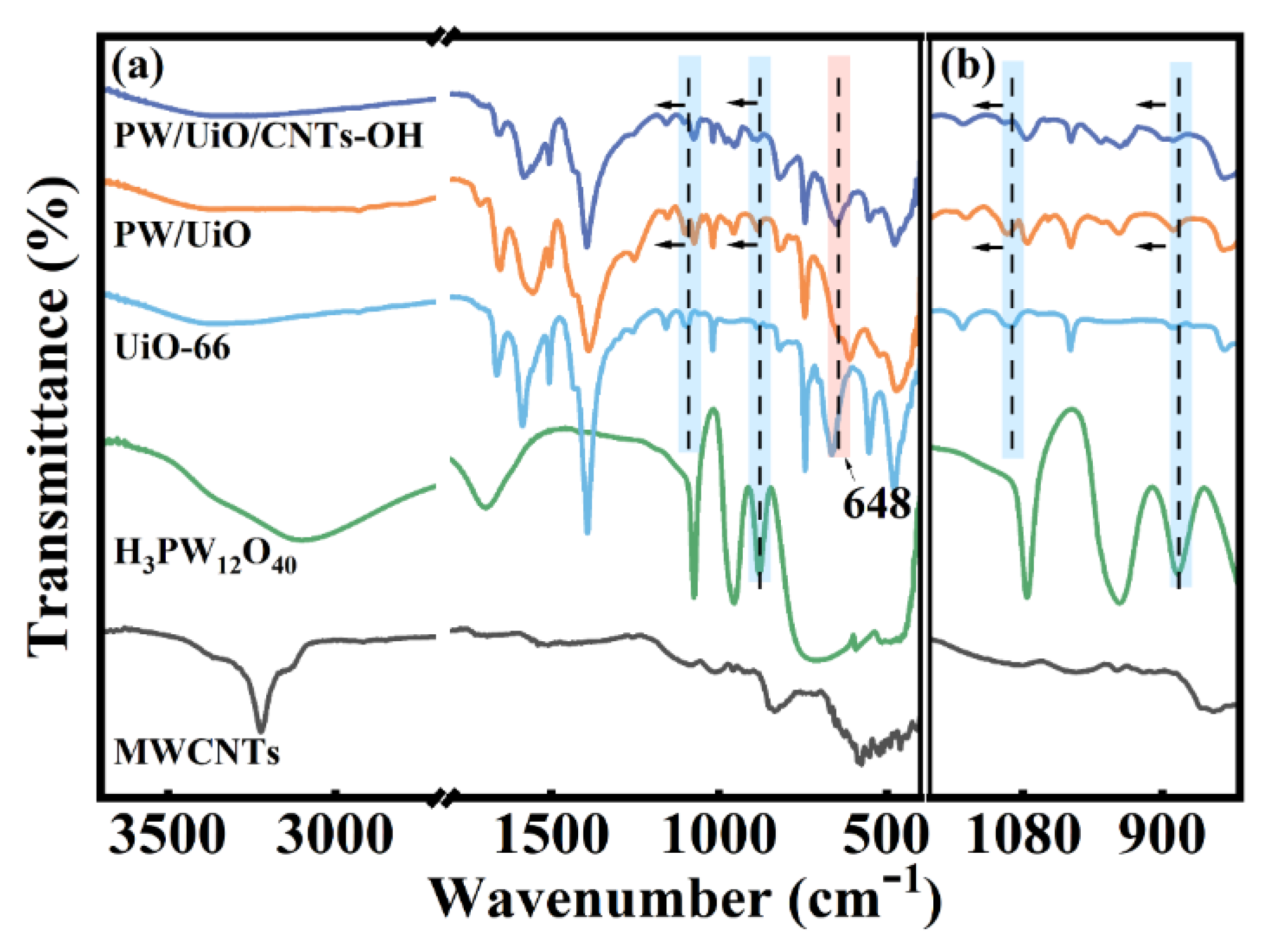
Figure 4.
(a)C 1s, (b)O 1s and (c)Zr 3d spectra of MWCNTs-OH, Zr-CNTs-OH, UiO/CNTs-OH, PW/UiO/CNTs-OH, (d)W 4f spectra of PW/UiO/CNTs-OH.
Figure 4.
(a)C 1s, (b)O 1s and (c)Zr 3d spectra of MWCNTs-OH, Zr-CNTs-OH, UiO/CNTs-OH, PW/UiO/CNTs-OH, (d)W 4f spectra of PW/UiO/CNTs-OH.
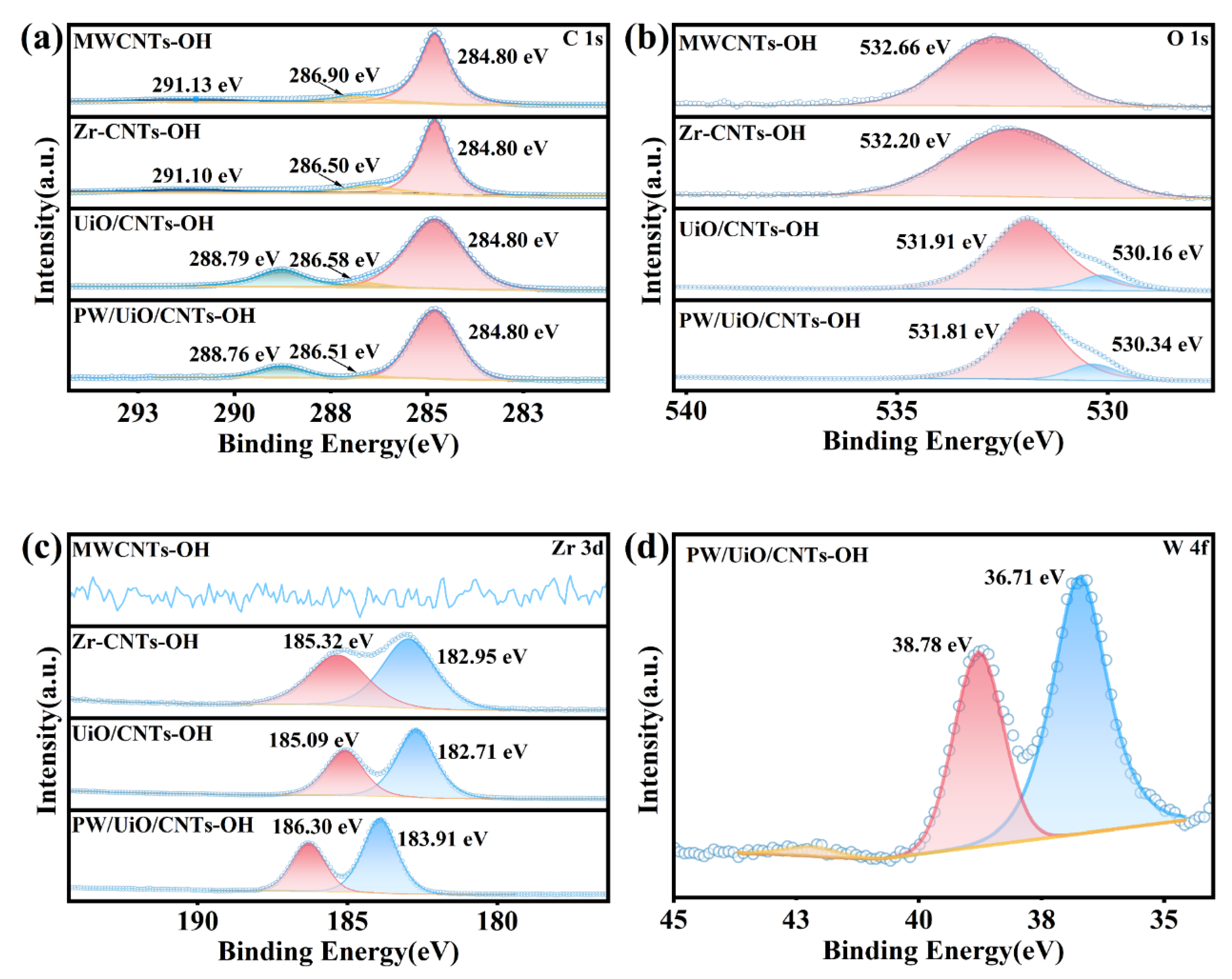
Figure 5.
The N2 adsorption-desorption isotherms and pore size distribution of MWCNTs-OH (a), UiO-66 (b), PW/UiO (c) and PW/UiO/CNTs-OH (d)
Figure 5.
The N2 adsorption-desorption isotherms and pore size distribution of MWCNTs-OH (a), UiO-66 (b), PW/UiO (c) and PW/UiO/CNTs-OH (d)
Figure 6.
NH3-TPD profile of (a) UiO-66, PW/UiO and PW/UiO/CNTs-OH, FT-IR spectra of (b) UiO-66, PW/UiO and PW/UiO/CNTs-OH, EIS plots of (c) UiO-66, PW/UiO and PW/UiO/CNTs-OH, TG curves of MWCNTs, UiO-66, PW/UiO and PW/UiO/CNTs-OH catalysts.
Figure 6.
NH3-TPD profile of (a) UiO-66, PW/UiO and PW/UiO/CNTs-OH, FT-IR spectra of (b) UiO-66, PW/UiO and PW/UiO/CNTs-OH, EIS plots of (c) UiO-66, PW/UiO and PW/UiO/CNTs-OH, TG curves of MWCNTs, UiO-66, PW/UiO and PW/UiO/CNTs-OH catalysts.
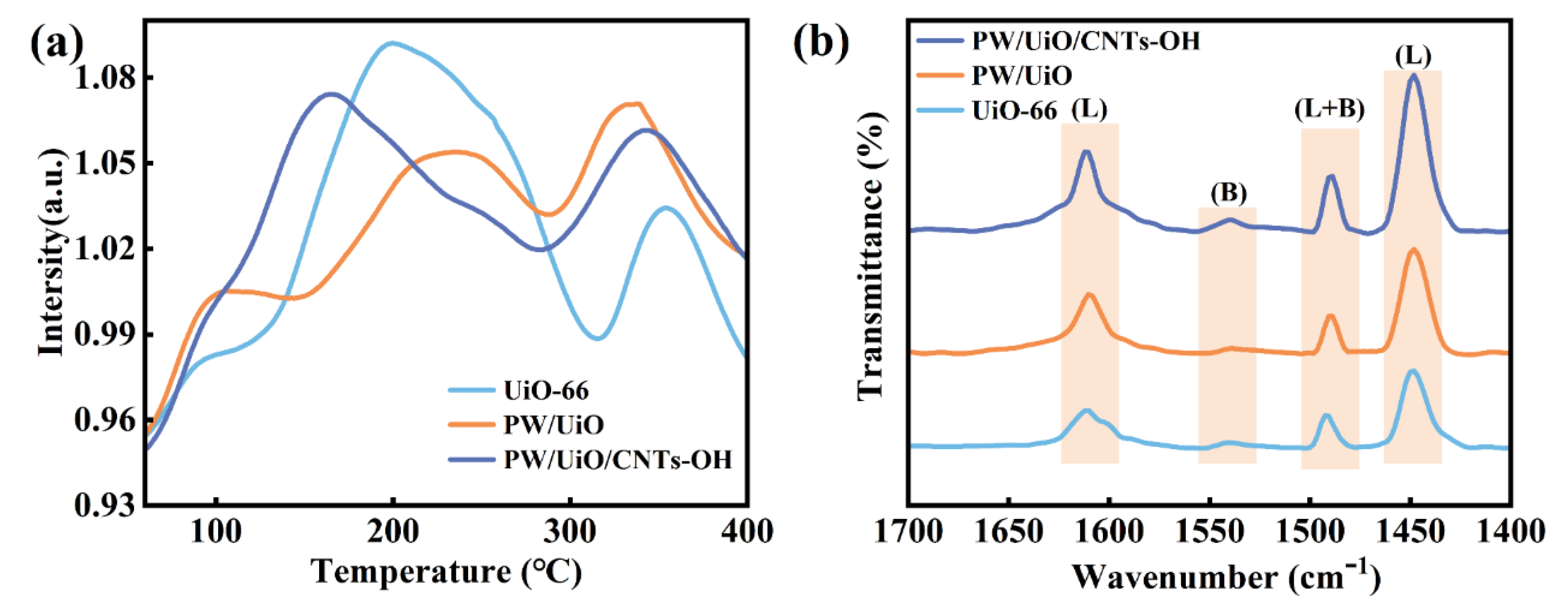
Figure 7.
The bridging role of UiO-66 in PW/UiO/CNTs-OH and the formation mechanism of Lewis acid sites
Figure 7.
The bridging role of UiO-66 in PW/UiO/CNTs-OH and the formation mechanism of Lewis acid sites
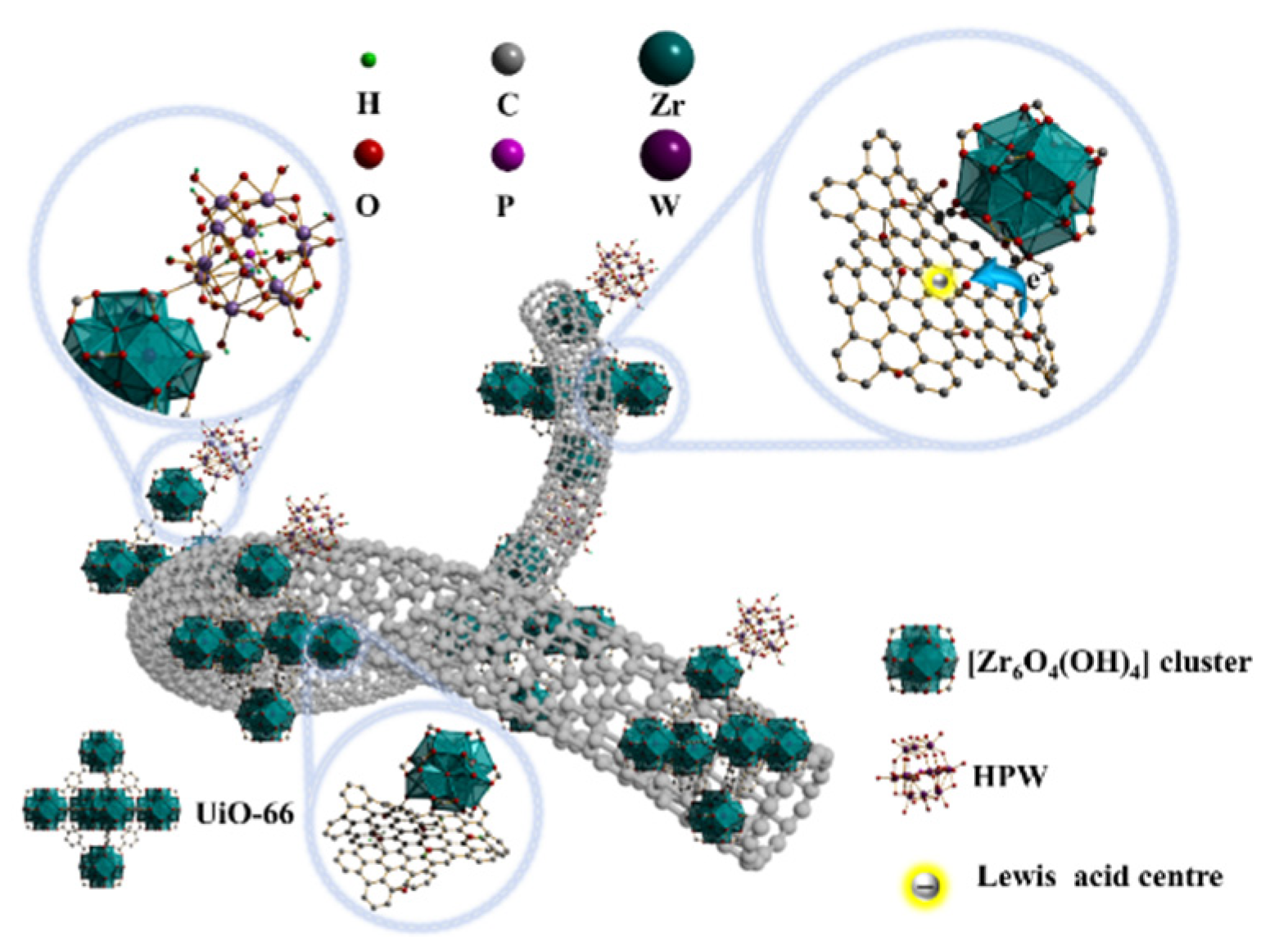
Figure 8.
The yield of FAME catalyzed by UiO-66, PW/UiO和PW/UiO/CNTs-OH
Figure 9.
Combination effect of (a) the molar ratio methanol to oleic acid and catalyst loading, (b) the molar ratio methanol to oleic acid and temperature, (c) catalyst loading and temperature on biodiesel yield.
Figure 9.
Combination effect of (a) the molar ratio methanol to oleic acid and catalyst loading, (b) the molar ratio methanol to oleic acid and temperature, (c) catalyst loading and temperature on biodiesel yield.
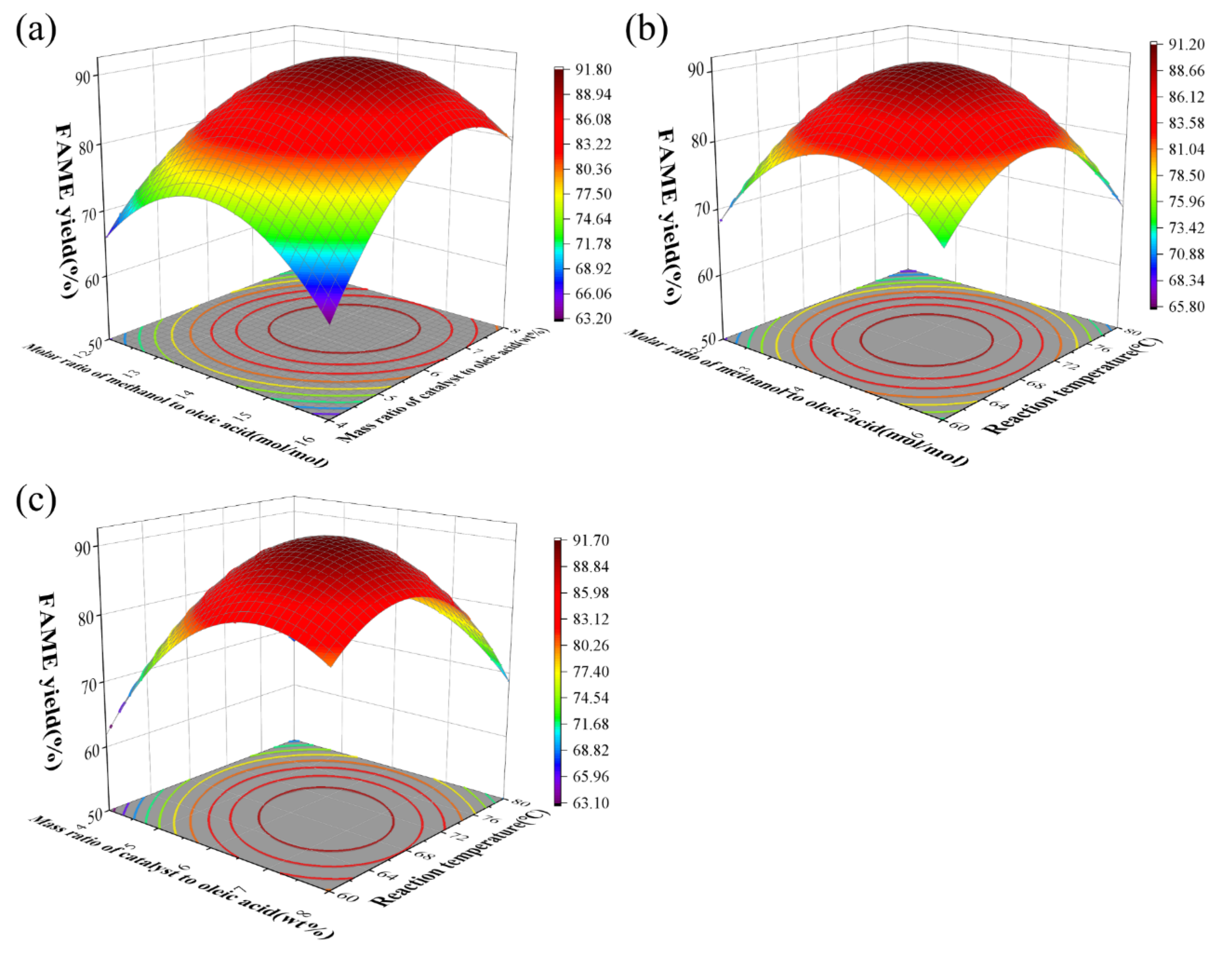
Figure 10.
The yield of FAME catalyzed by PW/UiO/CNTs-OH in five reaction cycles.
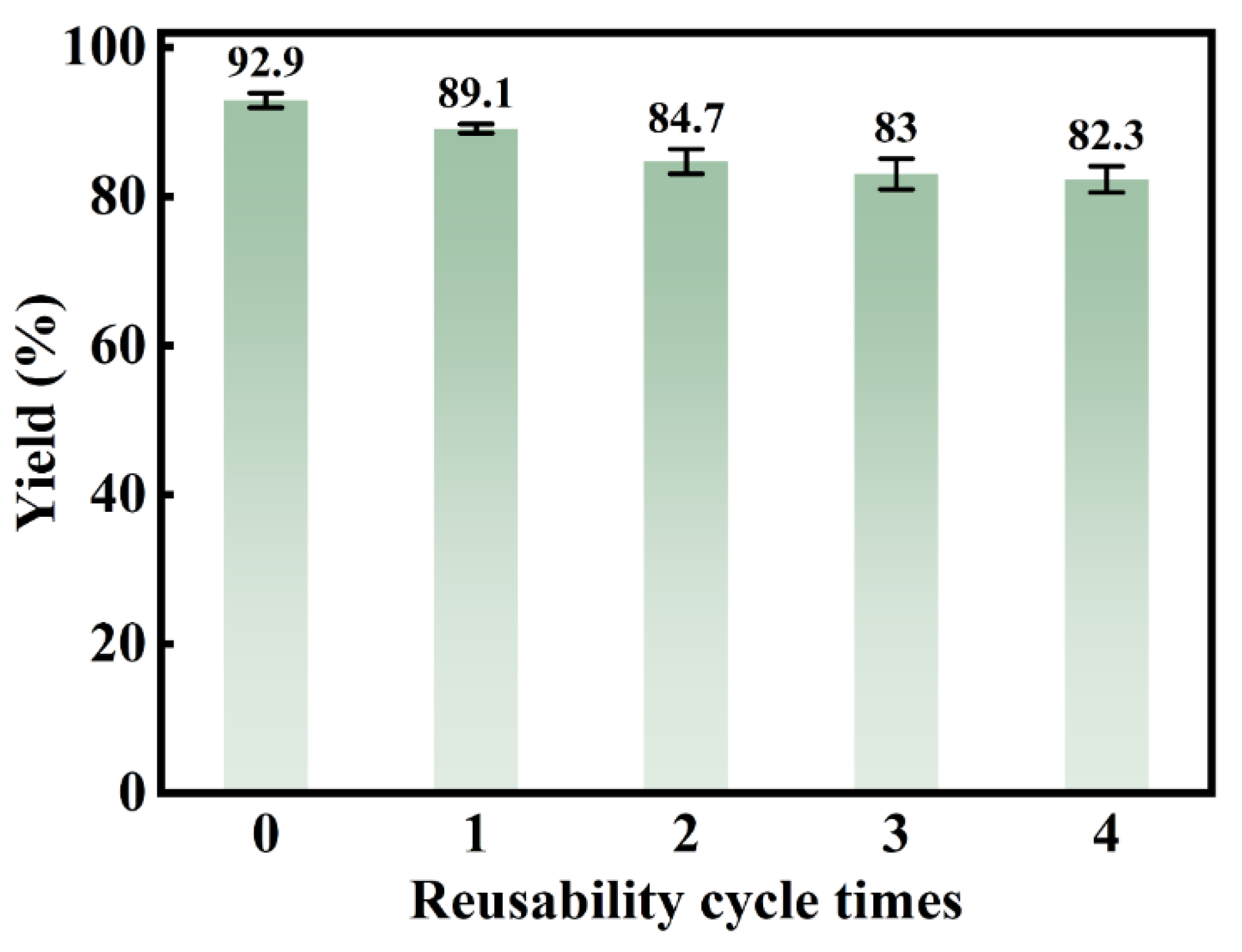

Table 1.
Range and levels of each process parameter based on the CCD-RSM methodology.
| Factor | Code | Range and Levels | ||||
| -ɑ (-1.682) | -1 | 0 | 1 | ɑ (1.682) | ||
| the molar ratio of methanol to oleic acid (mol/mol) | X1 | 10.64 | 12 | 14 | 16 | 17.36 |
| Catalyst loading (wt%) | X2 | 2.64 | 4 | 6 | 8 | 9.36 |
| Reaction temperature (°C) | X3 | 53.18 | 60 | 70 | 80 | 86.82 |
Table 2.
The actual content of W element in different catalysts
| Catalyst | Average mass of W (g/g of catalyst) | The actual W loading (wt%) |
|---|---|---|
| PW/CNTs | 0.0149 | 1.49 |
| PW/CNTs-OH | 0.0167 | 1.67 |
| PW/UiO-66 | 0.0439 | 4.39 |
| PW/UiO/CNTs-OH | 0.1487 | 14.87 |
Table 3.
Pore volume, pore diameter, and surface area analysis of the catalysts.
| Catalyst | Mean pore size (m2/g) | Pore volume (cm3/g) | Mean pore size (nm) |
|---|---|---|---|
| HPW | 2.830 | 0.005 | 7.178 |
| MWCNTs-OH | 163.590 | 1.347 | 32.939 |
| UiO-66 | 997.531 | 0.547 | 2.197 |
| PW/UiO | 700.046 | 0.555 | 3.173 |
| PW/UiO/CNTs-OH | 857.147 | 0.517 | 2.411 |
Table 4.
Acid content and acidic distribution of the catalysts
| Catalyst | Weak acidic site (mmol/g) | Moderate acidic site (mmol/g) | Brӧnsted acidity (μmol/g) | Lewis acidity (μmol/g) | Total acidity (mmol/g) | Brönsted/Lewis acidity ratio(B/L) |
|---|---|---|---|---|---|---|
| UiO-66 | 4.57 | 1.23 | 3.63 | 34.72 | 5.79 | 0.10 |
| PW/UiO | 3.64 | 2.58 | 3.65 | 44.54 | 6.21 | 0.08 |
| PW/UiO/CNTs-OH | 4.36 | 3.05 | 9.98 | 83.69 | 7.40 | 0.12 |
Table 6.
Results of FAME yield from the experimental design.
| Std. | X1 (mol/mol) | X2 (wt%) | X3 (°C) | Y (%) |
|---|---|---|---|---|
| 1 | 14 | 6 | 70 | 90.3 |
| 2 | 14 | 6 | 70 | 90.8 |
| 3 | 12 | 8 | 80 | 52.1 |
| 4 | 17.3636 | 6 | 70 | 62.4 |
| 5 | 14 | 2.6364 | 70 | 52.3 |
| 6 | 10.6364 | 6 | 70 | 55.8 |
| 7 | 16 | 8 | 60 | 73.8 |
| 8 | 12 | 4 | 60 | 50.3 |
| 9 | 12 | 4 | 80 | 59.9 |
| 10 | 14 | 6 | 70 | 90.8 |
| 11 | 16 | 4 | 60 | 51.1 |
| 12 | 14 | 9.3636 | 70 | 68.8 |
| 13 | 16 | 4 | 80 | 54.1 |
| 14 | 14 | 6 | 70 | 91.4 |
| 15 | 14 | 6 | 70 | 92.0 |
| 16 | 16 | 8 | 80 | 63.7 |
| 17 | 12 | 8 | 60 | 63.7 |
| 18 | 14 | 6 | 86.8179 | 57.2 |
| 19 | 14 | 6 | 53.1821 | 62.8 |
| 20 | 14 | 6 | 70 | 90.8 |
Table 7.
ANOVA for response surface quadratic model.
| Source | Sum of squares | Degree of freedom | Factor | F-value | p-value |
|---|---|---|---|---|---|
| Model | 4910.43 | 9 | 545.60 | 545.60 | < 0.0001 |
| X1 | 56.59 | 1 | 56.59 | 56.59 | < 0.0001 |
| X2 | 315.58 | 1 | 315.58 | 315.58 | < 0.0001 |
| X3 | 25.11 | 1 | 25.11 | 25.11 | 0.0011 |
| X1X2 | 89.11 | 1 | 89.11 | 89.11 | < 0.0001 |
| X1X3 | 3.25 | 1 | 3.25 | 3.25 | 0.1330 |
| X2X3 | 147.06 | 1 | 147.06 | 147.06 | < 0.0001 |
| X12 | 1794.60 | 1 | 1794.60 | 1794.60 | < 0.0001 |
| X22 | 1633.50 | 1 | 1633.50 | 1633.50 | < 0.0001 |
| X32 | 1693.71 | 1 | 1693.71 | 1693.71 | < 0.0001 |
| Residual | 12.16 | 10 | 1.22 | ||
| Lack of Fit | 10.39 | 5 | 2.08 | 5.88 | 0.0372 |
| Pure Error | 1.77 | 5 | 0.3537 | ||
| Total sum of squares | 4922.59 | 19 | |||
| Sted.Dev | 1.10 | R2 | 0.9975 | ||
| mean | 68.70 | Adj R2 | 0.9953 | ||
| C.V.% | 1.60 | Pred R2 | 0.9808 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



