
Submitted:
28 March 2025
Posted:
31 March 2025
You are already at the latest version
Abstract
We explored how the phosphorylation state of collapsin response mediator protein 2 (CRMP2) influences mitochondrial functions in cultured cortical neurons and cortical synaptic mitochondria isolated from APP-SAA KI mice, a knock-in APP mouse model of Alzheimer’s disease (AD). CRMP2 phosphorylation was increased at Thr 509/514 and Ser 522 in brain cortical lysates and cultured neurons from AD mice. The basal and maximal respiration of AD neurons were decreased. Mitochondria were hyperpolarized and superoxide anion production was increased in neurons from AD mice. In isolated synaptic AD mitochondria, ADP-stimulated and DNP-stimulated respiration were decreased, whereas the ADP-induced mitochondrial depolarization was reduced and prolonged. We found that CRMP2 binds to the adenine nucleotide translocase (ANT) in a phosphorylation-dependent manner. The increased CRMP2 phosphorylation in AD mice correlated with CRMP2 dissociation from the ANT and decreased ANT activity in AD mitochondria. On the other hand, recombinant CRMP2 (rCRMP2), added to the ANT-reconstituted proteoliposomes, increased ANT activity. A small molecule (S)-lacosamide ((S)-LCM), which binds to CRMP2 and suppresses CRMP2 phosphorylation by Cdk5 and GSK-3β, prevented CRMP2 hyperphosphorylation, rescued CRMP2 binding to the ANT, improved ANT activity, and restored mitochondrial membrane potential and respiratory responses to ADP and 2,4-dinitrophenol. Thus, our study highlights an important role for CRMP2 in regulating mitochondrial oxidative metabolism in AD by modulating ANT activity in a phosphorylation-dependent manner.
Keywords:
CRMP2
; Alzheimer’s disease
; cortical neurons
; mitochondrial oxidative metabolism
; mitochondrial ROS production
; adenine nucleotide translocase
1. Introduction
Alzheimer’s disease (AD) is a devastating, incurable neuropathology associated with neuronal dysfunction and memory impairment. AD is a frequent cause of dementia and a major contributor to illness and death in elderly individuals. One of the earliest neuropathological changes in AD is neuron degeneration, likely due to the deposition of β-amyloid plaques and neurofibrillary tangles in the brain. Neural degeneration, including neuronal dysfunction and loss of synapses, can contribute to cognitive and memory deficits. The mechanisms leading to neuronal dysfunction and synaptic defects in AD are not entirely clear.
Mitochondrial defects are a significant factor in AD pathology [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Mitochondrial oxidative metabolism plays a crucial role in satisfying energy demands in neurons [17,18,19]. Altered mitochondrial bioenergetics and mitochondrial morphological abnormalities have been reported in AD and have been proposed to contribute to synaptic dysfunction and neuronal degeneration [2,3,4,5,11,20,21,22]. However, the exact mechanisms leading to mitochondrial bioenergetic defects in AD are not completely understood.
Mitochondrial bioenergetics relies on the oxidative phosphorylation (OXPHOS) system. Mitochondrial F1F0-ATP synthase (ATP synthase) is the major component of the OXPHOS system [23], while adenine nucleotide translocase (ANT) is a key transporter in the inner mitochondrial membrane (IMM), exchanging cytosolic ADP for mitochondrial ATP [24,25]. The activities of ATP synthase and the ANT are reduced in AD [26,27,28,29,30], but the mechanisms contributing to this reduction are not entirely clear.
Collapsin response mediator protein 2 (CRMP2), is an abundant cytosolic phosphoprotein originally implicated in regulation of neurite outgrowth [31] and later in AD pathology [32,33,34,35,36]. CRMP2 is a physiological substrate for glycogen synthase kinase-3β (GSK-3β) and cyclin dependent kinase 5 (Cdk5), two protein kinases with increased activity in AD [37,38,39,40]. CRMP2 phosphorylation at the residues targeted by GSK-3β and Cdk5 is higher in human AD brains [41,42,43,44,45] and in the brains of mouse models of AD [43,45,46]. In AD mice, CRMP2 hyperphosphorylation was reported as early as 2 month of age and, thus, occurs prior to pathology, suggesting that increased CRMP2 phosphorylation is an early event in AD progression [43]. However, the functional consequences of CRMP2 hyperphosphorylation and their importance for AD pathology have not been investigated in detail.
CRMP2 binds to mitochondria [47,48]. A fraction of CRMP2 resides in the intermembrane space between the inner and the outer mitochondrial membranes [48], and interacts with the adenine nucleotide translocase 1 [47], a key player in mitochondrial bioenergetics [24]. CRMP2 hyperphosphorylation is paralleled by dissociation of CRMP2 from the ANT [47]. CRMP2 hyperphosphorylation in AD [41,42,43,44,45,46] implies that, in AD, CRMP2 may dissociate from the ANT. The consequences of CRMP2 dissociation from the ANT for mitochondrial oxidative metabolism in AD are not clear. It is also unclear whether preventing CRMP2 hyperphosphorylation and preserving CRMP2 interaction with the ANT are beneficial for mitochondrial oxidative metabolism.
In the present study, we investigated the effect of CRMP2 phosphorylation state on mitochondrial oxidative metabolism in APP-SAA KI mice, a knock-in AD mouse model [50]. We found increased CRMP2 phosphorylation in brain tissue lysates and in cultured cortical neurons from APP-SAA KI mice, which correlated with diminished CRMP2 binding to the ANT, decreased ANT activity, and altered mitochondrial respiration. The small molecule (S)-lacosamide ((S)-LCM) prevented CRMP2 hyperphosphorylation, restored CRMP2 binding to the ANT, rescued ANT activity, and improved respiration. Thus, in APP-SAA KI mice, CRMP2 affects mitochondrial oxidative metabolism in a phosphorylation-dependent manner by modulating ANT activity.
2. Materials and Methods
2.1. Animals
All manipulations with animals were accomplished in accordance with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals as well as in compliance with the Indiana University School of Medicine Institutional Animal Care and Use Committee approved protocol (#23156 MD/R/E). In our study, we used knock-in APP-SAA KI [50] (Jackson Laboratories, Strain # 034711), which carry humanized Aβ region R684H, F681Y, and G676R mutations, and the KM670/671NL (Swedish) mutation in exon 16 as well as the E693G (Arctic) and T714I (Austrian) mutations in exon 17 of the mouse App gene. As a control, we used B6J hAbeta mice (B6.Cg-Appem1Adiuj/J, Strain #033013, Jackson Laboratory), which express APP with a humanized Aβ 1-42 region as in APP-SAA KI mice, but without any mutations. With both strains mice of both sexes were used. Breeding colonies were maintained in the Laboratory Animal Resource Center at Indiana University School of Medicine, Indianapolis, IN. The APP-SAA KI mice are less prone to artifacts associated with overexpression of APP in transgenic mouse models of AD [50] and, therefore, in our experiments we were focused on APP-SAA KI mice.
2.2. Mouse Oral Gavage
We used mouse oral gavage to deliver a vehicle (10 μl DMSO in 0.2 ml saline) or (S)-LCM dissolved in DMSO (10 mg/kg body weight in 0.2 ml saline). A half milliliter (ml) syringe with a specialized 20-gauge needle for mouse oral gavage (FN-7910, Roboz Surgical Instrument Co., Gaithersburg, MD) were used.
2.3. Neuronal Cell Culture
Mouse cortical neurons in culture were prepared from postnatal day 1 (P1) APP-SAA KI and B6J hAbeta mice according to IACUC approved protocol and methodologies published earlier [51]. For immunoblotting and co-immunoprecipitation (co-IP) experiments neurons were grown at 200,000 cells per Petri dish (∅ 35 mm). For evaluation of mitochondrial membrane potential and superoxide anion production, neurons were grown at lesser density (10,000 cells) per glass bottom (∅ 10 mm) Petri dish to decrease likelihood of neuronal clumping. For all experiments with cells, 35 μg/ml uridine plus 15 μg/ml 5-fluoro-2’-deoxyuridine were injected to the dishes 24 hours after plating to inhibit propagation of microglia. Cells were kept in the air with 5% CO2 at 37°C in MEM with added 10% NuSerum (BD Bioscience, Bedford, MA) and 27 mM glucose. Neuronal cultures were utilized in experiments at 12-14 day in vitro (12-14 DIV).
2.4. Isolation and Purification of Brain Synaptic Mitochondria
2.5. Immunoblotting
Cultured cortical neurons (12-14 DIV) were prepared for gel-electrophoresis as follows. Cells were homogenized in a solution containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.1% SDS, 1 mM EDTA, and Phosphatase and Protease Inhibitors Cocktail (Roche, Cat # 04906845001 and Cat # 04693124001). The homogenates were incubated for 30 minutes on ice and then centrifuged at 100,000 g x 30 min. The pellet was discarded and the supernatant was utilized for gel-electrophoresis. Bis-Tris gels (4-12%, Invitrogen, Cat # NP0335) were used to separate proteins by electrophoresis (20 µg protein/lane). Then, proteins were translocated to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA, Cat # RPN78D). Blots were incubated at 22ºC for 60 minutes in a blocking solution. For phosphoprotein blotting, 5% BSA, Tris-HCl buffered saline, pH 7.2, plus 0.15% Triton X-100 was used. For total protein immunoblotting, 5% milk, phosphate-buffered saline, pH 7.2, plus 0.15% Triton X-100 was used. Following blocking, blots were incubated with either rabbit anti-CRMP2 pTyr 32 (a gift from Dr. Yoshio Goshima, Yokohama University Yokohama, Japan, 1:1500), sheep anti-CRMP2 pThr 509/514 (Kinasource, Cat # PB-043, 1:1500), rabbit anti-CRMP2 pSer 522 (ECM Biosciences, Cat # CP2191, 1:1500), rabbit anti-CRMP2 pThr 555 (ECM Biosciences, Cat # CP2251, 1:1500), rabbit anti-CRMP2 (Sigma, Cat # C2993, 1:1000), rabbit anti-ANT 1/2 antibody (Proteintech, Cat # 15997-1, 1:1000), anti-ATP5G1 (anti-subunit c of F1F0-ATP-synthase) rabbit monoclonal antibody (Boster Biological Technology, Pleasanton, CA, USA, Cat # M32382, 1:1000), or mouse anti-GAPDH (Abcam, Cat # ab9484, 1:2000) antibodies. Blots were then incubated with either goat anti-mouse or goat anti-rabbit IgG (1:25,000 or 1:20,000, respectively) coupled with horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and developed with Supersignal West Pico chemiluminescent reagents (Pierce, Rockford, IL, USA, Cat # 32106). Molecular mass marker Page Ruler Plus Prestained Protein Ladder (5μl, Thermo Fisher; Cat # 26619) was employed for determination of molecular masses of the bands. The immunoblot images were inverted, and the integrated density of bands was measured after background subtraction using Adobe Photoshop 22.2.0.
2.6. Co-Immunoprecipitation
We used Percoll gradient-purified brain cortical synaptic mitochondria isolated from 4-month old APP-SAA KI mice, untreated and treated with 10 mg/kg body weight (S)-LCM, delivered by oral gavage for 7 days prior to experiment or with a vehicle (10 μl DMSO in 0.2 ml saline delivered by gavage for 7 days prior to experiment). As a control, we used cortical synaptic mitochondria isolated from age-matched B6J hAbeta mice. After isolation, mitochondria were lysed in the buffer containing 125 mM KCl, 3 mM KH2PO4, 0.5 mM MgCl2, 10 mM Hepes, pH 7.4, Proteinase Inhibitor Cocktail (Roche), 1% NP40, and 0.1% SDS. Lysates were purified to eliminate any additional precipitate by treating with Protein A/G agarose beads (Santa Cruz Biotechnology, Cat # sc-2002, Santa Cruz, CA) for 2 hours at 4°C. Then, the lysates were stored overnight with primary rabbit anti-CRMP2 antibody (Sigma, Cat # C2993, 1:1000) or rabbit anti-ANT 1/2 antibody (Proteintech, Cat # 15997-1, 1:1000) under gentle shaking at 4°C followed by incubation with Protein A/G agarose beads (Santa Cruz Biotechnology, Cat # sc-2002) for 2 hours at 4ºC. In co-IP experiments with F1F0-ATP-synthase subunit c and CRMP2, we used anti-ATP5G1 (anti-subunit c of F1F0-ATP-synthase) rabbit monoclonal antibody (Boster Biological Technology, Pleasanton, CA, USA, Cat # M32382, 1:1000). The immune-captured complexes were washed 3 times with lysis buffer before being heated at 70°C in equal volumes of SDS loading dye (Invitrogen, Carlsbad, CA). In these experiments, Tris-Acetate gels (3–8%, Invitrogen, Cat # EA0375BOX) were employed to separate proteins by electrophoresis (20 µg protein/lane). Samples were subjected to immunoblotting as previously described [54,55]. Blots were probed with rabbit anti-CRMP2, rabbit anti-ANT 1/2, or rabbit anti-ATP5G1 antibody (each diluted 1:1000). All blots were produced at least in 3 independent experiments. The images of immunoblots were inverted and Integrated Density of bands was evaluated after background subtraction using Adobe Photoshop 22.2.0.
2.7. Cell Respirometry
A Seahorse XFe24 flux analyzer (Agilent Technologies, Santa Clara, CA, USA) was utilized to measure oxygen consumption rates (OCRs) of cultured cortical neurons (12 DIV) following the manufacturer’s recommendations. Neuronal cultures were plated in the 24-well assay plates at 105 cells per well. Prior to the experiment, the growth medium was exchanged for the standard bath solution supplemented with 10 mM glucose and 15 mM pyruvate. The standard bath solution contained 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, pH 7.4. The experiments were performed at 37ºC.
2.8. Mitochondrial Membrane Potential in Cultured Neurons
Mitochondrial membrane potential in cultured cortical neurons was evaluated with a fluorescent probe tetramethylrhodamine, methyl ester (TMRM, ThermoFisher Scientific, Cat # T668) [56]. Concurrently, cells were co-loaded with NeuroFluor™ NeuO as previously described [57]. After staining with NeuroFluor™ NeuO (Fisher Scientific, Cat # NC1363914), cells were loaded with 20 nM TMRM for 20 minutes at 37°C. TMRM (20 nM) was also added to the bath solution during experiments. Fluorescence of NeuroFluor™ NeuO was induced by illumination at excitation wavelength 480 ± 20 nm and recorded through a 505 nm dichroic mirror at emission wavelength 535 ± 25 nm. TMRM fluorescence was induced at excitation wavelength 545 ± 15 nm and recorded through a 565 nm dichroic mirror at emission wavelength 620 ± 30 nm. In experiments with TMRM, excitation of NeuroFluor™ NeuO at 545 ± 15 nm did not generate detectable fluorescence when we tried to record a signal through a 565 nm dichroic mirror at 620 ± 30 nm. Bright field and fluorescence images were taken with a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon CFI Plan Apo ×100 1.4 NA objective and CCD camera Cool SNAPHQ (Roper Scientific, Tucson, AZ, USA) controlled by MetaMorph 6.3 software (Molecular Devices, Downingtown, PA, USA).
2.9. Mitochondrial Superoxide Anion Production in Cultured Neurons
Mitochondrial superoxide anion production was assessed with MitoSOX Red (Molecular Probes) [58]. Simultaneously, cells were co-stained with NeuroFluor™ NeuO as described above. After loading with NeuroFluor™ NeuO, cells were loaded with 2.5 μM MitoSOX Red for 10 min at 37°C. Fluorescence of NeuroFluor™ NeuO was excited at 480 ± 20 nm and recorded through a 505 nm dichroic mirror at 535 ± 25 nm. Fluorescence of MitoSOX Red was excited at 545±15 nm and recorded through a 565 nm dichroic mirror at 620 ± 30 nm. In experiments with MitoTracker Red, TMRM, and MitoSOX Red, excitation of NeuroFluor™ NeuO at 545 ± 15 nm did not produce measurable fluorescence when we attempted to record through a 565-nm dichroic mirror at 620 ± 30 nm. Bright field and fluorescence images were acquired as described above.
2.10. Respiration and Membrane Potential in Isolated Mitochondria
Respiration and membrane potential of isolated brain cortical synaptic mitochondria were measured concurrently under uninterrupted stirring in a 0.4 ml chamber with a tightly sealed lid at 37ºC in the incubation medium that included 125 mM KCl, 3 mM KH2PO4, 0.5 mM MgCl2, 10 mM Hepes, pH 7.4, 0.1% BSA free from fatty acids, 10 μM EGTA, 1 mM malate and 3 mM pyruvate. The incubation chamber was outfitted with a miniature, home-made Clark-type oxygen electrode and a tetraphenylphosphonium (TPP+)-sensitive electrode. The slope of the oxygen electrode trace was used to calculate the respiratory rate. Mitochondrial membrane potential was assessed with a TPP+-sensitive electrode by following TPP+ distribution between the incubation medium and mitochondria [59]. An increase in TPP+ concentration outside of mitochondria indicated mitochondrial depolarization, while a decrease in TPP+ concentration outside of mitochondria indicated polarization of mitochondria.
2.11. Purification and Reconstitution of the ANT
Mitochondria were isolated from brains of ten C57BL/6J mice as we described previously [52,53]. The purification of the ANT from brain mitochondria was performed as described by Gawaz et al. [60] for the ANT from yeast mitochondria and for the ANT from bovine heart mitochondria as described by us [61]. The reconstitution of mouse brain ANT in proteoliposomes was performed as described earlier for the ANT from yeast and bovine heart mitochondria [60,61]. A mixture of 40 mg of phosphatidylcholine (Millipore-Sigma, Cat # 3356) and 1.5 mg of cardiolipin (Millipore-Sigma, Cat # C0563) was dissolved with 0.45 ml of a solution containing 11% detergent C12E8 (w/v, Millipore-Sigma, Cat # P8925), 87 mM Na2SO4, 1 mM EGTA, 175 mM Tricine-OH, pH 8.0, and 87 mM ATP. 0.45 ml of the dissolved lipids were mixed with 1.5 ml of the ANT extract at a final weight ratio of 0.015 protein/phospholipid. This gave a final concentration of 2.5% C12E8 (w/v), 20 mM Na2SO4, 0.23 mM EGTA, 40.4 mM Tricine-OH, pH 8.0, 9.6 mg/ml phospholipid, and 20 mM ATP. Proteoliposomes were formed by slow removal of C12E8 with ion exchange beads Amberlite XAD-4 (Millipore-Sigma, Cat # XAD4). For the removal of the external solution, the proteoliposomes were passed through Sephadex G-75 (Millipore-Sigma, Cat # G75120) column (30x1 cm) pre-equilibrated with 100 mM sucrose, 30 mM Na2SO4, 1 mM Tricine-OH, pH 7.5, 1 mM EDTA.
2.12. Preparation of Recombinant CRMP2 (rCRMP2)
To produce recombinant CRMP2 protein, we followed established protocols [62]. We transformed BL21 (DE3) Escherichia coli with DNA encoding sequence-verified pGex-Glu-CaV2.2-type channel constructs for protein expression. Dr. Akihiro Kurimasa from Tottori, Japan, kindly provided the CRMP-2-GST fusion constructs. Protein expression was initiated using 1 mM isopropyl-β-d-thiogalactopyranoside. For purification, the transformed bacteria were grown overnight at 16°C, then pelleted and lysed in a buffer containing 20 mM Tris (pH 7.5), 200 mM NaCl, 0.1 mM EDTA, 1 mM dithiothreitol, and protease inhibitors. This was done using an M-110L microfluidizer from Microfluidics Corp., Newton, MA. The lysate was then treated with Triton X-100 (1% final concentration) and incubated on ice for 30 minutes. Finally, the mixture was centrifuged at 30,000 × g for 45 minutes at 4°C. The pure CRMP2 protein was concentrated to 30 mg/mL, flash frozen in liquid nitrogen and stored at −80°C in buffer containing 25 mM Tris-HCl, 100 mM glycine, pH 7.3, 10% glycerol [63].
2.13. Evaluation of Adenine Nucleotide Translocase activity
Percoll gradient-purified brain cortical synaptic mitochondria (30 μg protein) were incubated at 37ºC in 0.4 ml of incubation medium containing 125 mM KCl, 0.5 mM MgCl2, 3 mM KH2 PO4, 10 mM Hepes, pH 7.4, 0.1% BSA free from fatty acids, 10 μM EGTA, 3 mM pyruvate plus 1 mM malate. The activity of the ANT was evaluated by following ATP efflux from mitochondria in the incubation medium, induced by adding 100 μM ADP as described previously [29,64] with some modifications. ATP in the incubation medium was assessed using the coupled ATP detecting enzymatic system consisting of 2.5 mM glucose, 1.0 U/ml hexokinase (Millipore-Sigma, Cat # H4502), 1.0 U/ml glucose-6-phosphate dehydrogenase (Millipore-Sigma, Cat # G6378) and 0.5 mM NADP+ (Millipore-Sigma, Cat # NADP-RO) in the presence of 10 μM P1,P5-Di(adenosine-5′) pentaphosphate (Ap5A, Millipore-Sigma, Cat # D4022), a specific inhibitor of adenylate kinase [65]. The rate of NADP+ reduction and formation of NADPH in the incubation medium is stoichiometrically equivalent to ATP release from mitochondria in the incubation medium mediated by the ANT [29,64]. The formation of NADPH in the incubation medium was followed by measuring NADPH fluorescence (excitation/emission wavelengths 340/460 nm) under gentle stirring with Perkin-Elmer LS 55 luminescence spectrometer equipped with a bio-kinetics accessory. The rate of NADPH formation was determined using the tangent to the initial fragment of the NADPH fluorescence trace and expressed as nmol NADPH x min-1 x mg protein-1. Control experiments were carried out in the presence of 5 μM carboxyatractyloside (CAT, Millipore-Sigma, Cat # TA9H93ED6E11) and 5 μM bongkrekic acid (BKA, Millipore-Sigma, Cat # B6179), the specific ANT inhibitors [24,66,67], to confirm that the ATP release was solely mediated by the ANT.
In experiments with ANT-reconstituted proteoliposomes, 40 μl of proteoliposomes were added to 360 μl of medium containing 100 mM sucrose, 30 mM Na2SO4, 1 mM Tricine-OH, pH 7.5, 1 mM EDTA. The ADP/ATP exchange was initiated by adding 100 μM ADP. In some experiments, the ANT proteoliposomes were pretreated with 10 μg/ml of recombinant CRMP2 (rCRMP2) for 5 minutes under gentle stirring prior to adding ADP. The ATP release from proteoliposomes was measured as described above, but without Ap5A.
2.14. Statistics
The experimental results are shown as mean ± SD of the specified number of individual experiments. Statistical analysis of the experimental data included unpaired t-test or one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test (GraphPad Prism® version 4.0, GraphPad Software Inc., La Jolla, CA, USA). Each experiment was conducted using several different preparations of isolated mitochondria or cultured neurons.
3. Results
3.1. CRMP2 Phosphorylation
Previously, it was reported that CRMP2 is hyperphosphorylated at Thr 509/514 and Ser 522 in cultured cortical neurons derived from transgenic APP/PS1 mice [68] and in postmortem brain tissues of AD patients [43,44,45,68]. In the present experiments, we found that CRMP2 was hyperphosphorylated at Thr 509/514 and Ser 522, in lysates of brain cortices of 4-month old APP-SAA KI mice (Figure 1). The unedited images of immunoblots are shown in Suppl. Figure S1.
We also found that CRMP2 was hyperphosphorylated at Tyr 32, Thr 509/514, Ser 522, and Thr 555 in cultured cortical neurons from APP-SAA KI mice (Figure 2).
The unedited images of immunoblots are shown in Suppl. Figure S2. To attenuate CRMP2 phosphorylation, we used the small molecule, (S)-LCM. We had previously demonstrated that (S)-LCM binds to CRMP2 and prevents CRMP2 hyperphosphorylation by Cdk5 and GSK-3β kinases, without inhibiting these kinases [69,70]. (S)-LCM delivered to mice by oral gavage (10 mg/kg body weight for 7 days) attenuated CRMP2 phosphorylation in brain cortices of APP-SAA KI mice (Figure 1). In addition, (S)-LCM (10µM in the growth medium for 7 days prior to experiment) applied to cultured neurons from APP-SAA KI mice reduced CRMP2 phosphorylation at Thr 509/514 and Ser 522, but not at Tyr 32 and Thr 555 (Figure 2).
3.2. Cell Respirometry
To evaluate the functional consequences accompanying alterations in CRMP2 phosphorylation state, we used cultured cortical neurons from APP-SAA KI and B6J hAbeta mice and subjected them to cell respirometry with the Seahorse XFe24 analyzer. In neurons from APP-SAA KI mice, we found a reduction in the rates of basal and maximal respiration, stimulated by the uncoupler 2,4-dinitrophenol (DNP), compared with respiration of neurons from B6J hAbeta mice (Figure 3). Both the basal and the maximal respiration of APP-SAA KI neurons were restored by pre-treatment of cells with 10µM (S)-LCM for 7 days prior to experiment (Figure 3).
3.3. Mitochondrial Membrane Potential and Superoxide Production in Cultured Neurons
The decrease in respiration (Figure 3) could be due to inhibition of the electron transport chain or due to inhibition of oxidative phosphorylation (OXPHOS) system. If the electron transport chain is inhibited, mitochondria should be depolarized; if the OXPHOS system is inhibited, mitochondria should be hyperpolarized. To clarify this, we used TMRM to evaluate mitochondrial membrane potential in cultured cortical neurons from APP-SAA KI and B6J hAbeta mice. Mitochondria in cortical neurons from APP-SAA KI mice were hyperpolarized (Figure 4), consistent with previous findings with mitochondria of APPNL-G-F/NL-G-F mice, another knock-in AD mouse model [71]. Hyperpolarization of APP-SAA KI mitochondria is consistent with reduced basal respiration of APP-SAA KI neurons (Figure 3) and is most likely due to suppression of OXPHOS system as a result of inhibition of the ANT, or F1F0-ATP synthase, or both.
We also found a higher level of superoxide anion (O2-) production in mitochondria of cortical neurons from APP-SAA KI mice (Figure 5), in line with previous reports of elevated ROS production in mitochondria of knock-in APPNL-G-F/NL-G-F mice [71]. The high O2- production in AD mitochondria could be due to elevated mitochondrial membrane potential [72] (Figure 4) and could facilitate an induction of the detrimental permeability transition pore (PTP) in AD mitochondria [73,74,75]. In addition, elevated ROS levels could oxidize CRMP2 and enhance its phosphorylation by GSK-3 [76], creating a self-perpetuating vicious cycle of CRMP2 oxidation and phosphorylation.
3.4. Respiration and Membrane Potential in Isolated Mitochondria
To untangle the mechanisms underlying the decline in the basal and the maximal respiration in APP-SAA KI neurons, we measured oxygen consumption rates and mitochondrial membrane potential in isolated brain cortical synaptic mitochondria (Figure 6). Mitochondria from APP-SAA KI mice accumulated similar amounts of lipophilic cation tetraphenylphosphonium (TPP+) compared with B6J hAbeta mitochondria, suggesting similar membrane potential in these mitochondria. Addition of ADP accelerated mitochondrial respiration and transiently depolarized mitochondria. In mitochondria from APP-SAA KI mice, ADP-stimulated respiration was slower, and depolarization was smaller and prolonged (Figure 6). The duration of ADP-induced depolarization, from onset to steady state after repolarization, was 189±6 seconds in APP-SAA KI mitochondria compared to 124±7 seconds in B6J hAbeta mitochondria (N=7, p<0.01).This suggests that either the F1F0-ATP synthase is inhibited in APP-SAA KI mitochondria as reported earlier [26,27], or ADP enters AD mitochondria slower due to inhibition of the ANT [28,29,30], or both. In addition to ADP-stimulated respiration, the maximal respiration stimulated by DNP was lower with APP-SAA KI mitochondria compared with B6J hAbeta mitochondria. Of note, DNP effect on mitochondrial respiration also depends on ANT activity [77], but not on F1F0-ATP synthase activity, hence rejecting the hypothesis about F1F0-ATP synthase involvement in respiratory alterations in AD mitochondria (Figure 6) and AD neurons (Figure 3).
(S)-LCM-attenuated CRMP2 phosphorylation in brain cortices of APP-SAA KI mice (Figure 1) correlated with improved mitochondrial response to ADP (duration of depolarization: 132.2±8 sec, N=7, p<0.01) and accelerated DNP-stimulated maximal respiration (Figure 6). Thus, the changes in respiration of isolated mitochondria are consistent with the changes in respiration of cultured neurons. It is conceivable that CRMP2, in a phosphorylation-dependent manner, modulates ANT-dependent changes in mitochondrial respiration and membrane potential. It is possible that this occurs due to CRMP2 interaction with the ANT and CRMP2-mediated modulation of ANT activity.
3.5. CRMP2-ANT Co-Immunoprecipitation
In the following experiments, we investigated plausible interaction of CRMP2 with the ANT. We detected CRMP2 interaction with the ANT using co-immunoprecipitation (co-IP) assay applied to cortical synaptic mitochondria isolated from the control, non-AD B6J hAbeta mice (Figure 7). This interaction was disrupted in APP-SAA KI mitochondria and rescued by (S)-LCM pre-treatment of APP-SAA KI mice (Figure 7). The unedited images of immunoblots are shown in Suppl. Figure S3. CRMP2 is hyperphosphorylated in cortices of APP-SAA KI mice, whereas (S)-LCM prevented CRMP2 hyperphosphorylation (Figure 1), suggesting that CRMP2 interacts with the ANT in a phosphorylation-dependent manner and possibly may modulate ANT activity. We also tested CRMP2 interaction with c-subunits of F1F0-ATP synthase, exposed to the intermembrane space between the outer and the inner mitochondrial membranes, but did not find evidence for this kind of interaction (Suppl. Figure S4). Therefore, we were focused on CRMP2-ANT interaction.
3.6. ANT Activity in Isolated Mitochondria
To evaluate the functional consequences of CRMP2 interaction with the ANT, we measured ANT transport activity in brain cortical synaptic mitochondria isolated from B6J hAbeta and APP-SAA KI mice. We used a previously described method [29,64] with some modifications. This method is based on NADP+ reduction and formation of NADPH in the coupled biochemical reactions catalyzed by hexokinase and glucose-6-phosphate dehydrogenase. In these reactions, formation of NADPH is stoichiometrically equivalent to ATP release from mitochondria, which is then used in these reactions. The ATP release from mitochondria via the ANT-mediated ADP/ATP exchange was initiated by adding 100 μM ADP to mitochondria. NADPH formation was monitored by following NADPH autofluorescence. The rate of NADPH formation, corresponding to the rate of ATP release from mitochondria, was determined by assessing the tangent to the initial fragment of the NADPH fluorescence signal following ADP addition. Using this method, we found a decrease in the ANT activity in mitochondria from APP-SAA KI mice compared with mitochondria from B6J hAbeta mice (Figure 8). (S)-LCM pre-treatment of APP-SAA KI mice prevented CRMP2 hyperphosphorylation (Figure 1), restored CRMP2-ANT interaction (Figure 7), and significantly increased the rate of ATP release (Figure 8), suggesting improved ANT activity. Carboxyatractyloside (CAT, 5 μM) and bongkrekic acid (BKA, 5 μM), specific inhibitors of the ANT [24,66,67], completely inhibited NADPH formation, reflecting suppression of ATP release and indicating that the ANT is solely responsible for the ATP transport. Thus, these results suggest that ANT activity is decreased in APP-SAA KI mitochondria and its activity is correlated with CRMP2 phosphorylation state and CRMP2 binding to the ANT.
3.7. ANT Activity in ANT-Reconstituted Proteoliposomes
An inhibition of F1F0-ATP synthase and reduced ATP production could also contribute to the decreased ATP release from APP-SAA KI mitochondria, measured with the NADP+ reduction assay [29,64], which we used in our experiments. Based on experiments with isolated mitochondria (Figure 8), we could not rule out this scenario completely. Thus, whether CRMP2 can directly affect ANT activity remained unclear. Consequently, to clarify this issue we tested whether CRMP2 selectively modulates ANT activity in experiments with the ANT-reconstituted proteoliposomes. The ANT was purified using hydroxyapatite chromatography and incorporated in the phospholipid liposomes as we described earlier [60,61]. The ANT-proteoliposomes were preloaded with 20 mM ATP. An addition of ADP (100μM) to ANT-proteoliposomes resulted in ATP release (Figure 9), measured as described above. Pretreatment of the ANT-proteoliposomes with recombinant CRMP2 (rCRMP2, 10 μg/ml for 5 minutes under gentle stirring prior to adding ADP) accelerated ATP release, whereas CAT (5 μM) and BKA (5 μM) completely inhibited ADP/ATP exchange. These results strongly suggest that CRMP2 stimulates ANT activity, whereas removal of CRMP2 from the ANT is associated with a decrease in ANT transport function.
4. Discussion
Alzheimer’s disease (AD) is associated with synaptic dysfunction and neuronal degradation [11,21,22]. Defects in mitochondrial dynamics and bioenergetics are major contributors to these alterations [1,2,3,4,5,6,7,8,9,10,11,12,13]. The mechanisms of mitochondrial defects in AD are not completely understood, but it is possible that CRMP2 is involved in modulation of mitochondrial functions in AD. Indeed, in our previous study we demonstrated that CRMP2 is involved in regulation of mitochondrial dynamics in AD [68]. CRMP2 binds to brain synaptic mitochondria [48,68], with a portion residing in the intermembrane space between the outer and inner mitochondrial membranes [48], where it may interact with various proteins, including the ANT [47].
CRMP2 is a cytosolic phosphoprotein that may influence activity and/or location of different proteins [32]. It is phosphorylated by GSK-3β and Cdk5 kinases [42,78,79,80], which are more active in AD [37,38,39,40]. Hyperphosphorylation of CRMP2 at specific residues is observed in AD brains [41,42,43,44,45] and AD mouse models (APP/PS1 and Tg2576 mice [43,45,46,81]), suggesting its involvement in early AD events [43]. Cortical neurons from wild-type E14 mice cultured for 3 days and then exposed to a toxic fragment of Aβ peptides (Aβ25–35) for another 3 days have a significant increase in CRMP2 phosphorylation [82], indicating that CRMP2 hyperphosphorylation in cultured cortical neurons may occur very early following a short exposure to Aβ25–35. Consequently, in our study we were focused on the newly discovered role of CRMP2 in regulating mitochondrial functions [48,49,68] and its relevance to AD pathology [68].
In this study, we used brain tissues, cultured cortical neurons, and cortical synaptic mitochondria isolated from APP-SAA KI mice, a knock-in mouse model of AD [50]. The APP-SAA KI mice carry humanized Aβ region R684H, F681Y, and G676R mutations, and the KM670/671NL (Swedish) mutation in exon 16 as well as the E693G (Arctic) and T714I (Austrian) mutations in exon 17 of the mouse App gene. At 4 months of age, Aβ deposition and increased Aβ42/Aβ40 ratio were detected in APP-SAA KI mice at levels comparable to Tg APP/PS1 mice [50]. The APP-SAA KI mice exhibited increased levels of biomarkers of neurodegeneration, suggesting neuronal degradation [50]. Possible alterations in mitochondrial oxidative metabolism have not been studied with these mice. However, mitochondrial defects were found in APPNL-G-F/NL-G-F mice, another knock-in APP mouse model [71]. These mice have decreased mitochondrial respiration, reduced ATP production, and increased mitochondrial membrane potential.
The precise mechanisms of CRMP2 involvement in AD pathology have not yet been delineated. In APP/PS1 mice, pioglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, decreased CRMP2 phosphorylation, improved energy metabolism, and mitigated motor coordination impairment and long term depression , but it is uncertain whether the effects were due to PPARγ activation or CRMP2 dephosphorylation [81,83]. Conversely, Aβ25–35 oligomers induced memory impairment and synaptic plasticity in wild-type mice, but not in phospho-CRMP2 deficient crmp2ki/ki mice [84], which lacked CRMP2 phosphorylation at Ser 522 and Thr 509/514 due to Ser 522 replacement with Ala [85]. These results suggest that preventing CRMP2 hyperphosphorylation by Cdk5 and GSK-3β, two kinases activated in AD [37,38,39,40], safeguards against the cognitive decline induced by Aβ25–35 oligomers [84]. Thus, CRMP2 hyperphosphorylation may play a crucial role in AD. However, the mechanisms by which CRMP2 contributes to AD pathology have not yet been explored.
In our study, we found that activity of the ANT is decreased in synaptic mitochondria from APP-SAA KI mice. In experiments with isolated cortical synaptic mitochondria, we found that the decrease in ANT activity correlated with CRMP2 hyperphosphorylation and CRMP2 dissociation from the ANT. (S)-LCM prevented CRMP2 hyperphosphorylation, restored CRMP2 interaction with ANT, and improved ATP transport across the IMM, allowing us to infer CRMP2-mediated modulation of ANT activity. The stimulating interaction of unphosphorylated CRMP2 with the ANT was further supported by our experiments with rCRMP2 and ANT-proteoliposomes. Importantly, the stimulating interaction of CRMP2 with the ANT might be not unique. Earlier, it was proposed that Bcl-2 interaction with the ANT could stimulate its translocator function, while interaction with pro-apoptotic protein Bax could diminish the translocator activity [86].
In our experiments, we used the NADP+ reduction assay to assess ANT activity in isolated cortical synaptic mitochondria [29,64]. In contrast to the originally reported method [29], in which the authors measured NADPH spectroscopically by following NADPH absorbance at 340 nm, we measured NADPH autofluorescence to determine the rate of ATP release from mitochondria. This approach appears to be more sensitive than measuring NADPH absorbance [87]. Although NADP+ reduction assay is a useful method to measure the rate of ATP efflux from mitochondria mediated by the ANT, it cannot completely distinguish between changes in ANT transport activity and alterations in F1F0-ATP synthase enzymatic activity. Indeed, the activities of F1F0-ATP synthase and the ANT are decreased in AD [26,27,28,29,30], but the relative significance and mechanisms contributing to these changes are not entirely clear. CRMP2 residing in the intermembrane space could potentially interact not only with the ANT, but also with c-subunits of F1F0-ATP synthase, facing the intermembrane space. However, in our co-IP experiments we did not find evidence for CRMP2 interaction with c-subunit of F1F0-ATP synthase. Nevertheless, the attenuation of CRMP2 phosphorylation and the enhanced interaction of CRMP2 with the ANT induced by (S)-LCM strongly correlated with the rate of ATP translocation from mitochondria. Thus, it is likely that in our experiments, CRMP2 modulated ANT activity rather than the activity of F1F0-ATP synthase.
This notion is also supported by measurements of neuronal respiration. Whereas the reduction in basal respiration could be due to suppression of ANT transport function or a decrease in F1F0-ATP synthase activity, the decrease in maximal DNP-stimulated respiration is independent of F1F0-ATP synthase activity, but depends on activity of the ANT [77]. Preventing CRMP2 hyperphosphorylation with (S)-LCM and restoring the CRMP2-ANT interaction effectively rescued both basal and maximal DNP-stimulated respiration, suggesting that CRMP2 regulates respiration by modulating ANT activity. Interestingly, despite the decreased maximal respiration of APP-SAA KI neurons being significantly higher than the basal respiration rate of control B6J hAbeta neurons, the sufficient respiratory reserve capacity suggests that this decrease likely does not account for the reduced basal respiration observed in APP-SAA KI mice.
Previous reports indicate that oxidative damage to ANT could reduce its transport activity in AD [29]. Additionally, inhibition of the mitochondrial voltage-dependent anion channel (VDAC) in AD may weaken ADP/ATP exchange between the mitochondrial matrix and the cytosol [30]. While these scenarios cannot be ruled out, they may not be the only mechanisms affecting ADP/ATP exchange in AD. Our data strongly suggest that CRMP2 hyperphosphorylation and its dissociation from ANT may contribute to ANT inactivation in AD. Therefore, our findings highlight the complexity and potential diversity of mechanisms affecting ANT and mitochondrial oxidative metabolism in AD.
In our previous study, we emphasized CRMP2’s role in regulating mitochondrial dynamics, including changes in morphology and motility, and its potential contribution to neuronal degradation in AD [68]. In this study, we demonstrated that CRMP2 binds to ANT in a phosphorylation-dependent manner and modulates mitochondrial oxidative metabolism. It is plausible that both alterations in mitochondrial dynamics and changes in oxidative metabolism contribute to mitochondrial defects and subsequent neuronal degeneration in AD. However, the extent to which these mechanisms contribute to neuronal degradation remains uncertain and will be the focus of our future studies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. The unedited images of immunoblots for Figure 1A. Figure S2. The unedited images of immunoblots for Figure 2A. Figure S3. The unedited images of immunoblots for Figure 7. Figure S4. The lack of evidence for CRMP2 interaction with c-subunits of F1F0-ATP synthase.
Author Contributions
T.B. performed experiments and analyzed data, R.K. analyzed data, provide critical components, and wrote the paper, N.B. conceived the project, performed experiments, analyzed data, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a National Institutes of Health NINDS grant R01 NS098772 to NB and RK. In addition, NB was supported by Faculty Research Support Program – External Resubmission grant from Indiana University, and Biomedical Research Grant from Indiana University School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Indiana University School of Medicine.
Institutional Review Board Statement
The animal study protocol was approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee (#23156 MD/R/E, 01/26/2024) for studies involving animals.
Informed Consent Statement
Not applicable
Data Availability Statement
Data are contained within the article or supplementary materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 14670-14675. [CrossRef]
- Selfridge, J.E.; E L; Lu, J.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis., 2013, 51, 3-12. [CrossRef]
- Martins, I.V.; Rivers-Auty, J.; Allan, S.M.; Lawrence, C.B. Mitochondrial Abnormalities and Synaptic Loss Underlie Memory Deficits Seen in Mouse Models of Obesity and Alzheimer’s Disease. J. Alzheimers. Dis., 2017, 55, 915-932. [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet., 2011, 20, 4515-4529.
- Flannery, P.J.; Trushina, E. Mitochondrial dynamics and transport in Alzheimer’s disease. Mol. Cell Neurosci., 2019, 98, 109-120. [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D. et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med., 2008, 14, 1097-1105. [CrossRef]
- Pedros, I.; Petrov, D.; Allgaier, M.; Sureda, F.; Barroso, E.; Beas-Zarate, C.; Auladell, C.; Pallas, M.; Vazquez-Carrera, M.; Casadesus, G. et al. Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim. Biophys. Acta, 2014, 1842, 1556-1566. [CrossRef]
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. Alzheimer’s disease: diverse aspects of mitochondrial malfunctioning. Int. J. Clin. Exp. Pathol., 2010, 3, 570-581.
- Cenini, G.; Voos, W. Mitochondria as Potential Targets in Alzheimer Disease Therapy: An Update. Front Pharmacol., 2019, 10, 902. [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers. Dis., 2018, 62, 1403-1416. [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers. Dement., 2023, 19, 333-342. [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener., 2020, 15, 30. [CrossRef]
- Cadonic, C.; Sabbir, M.G.; Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol. Neurobiol., 2016, 53, 6078-6090. [CrossRef]
- Parodi-Rullan, R.; Sone, J.Y.; Fossati, S. Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Alzheimers. Dis., 2019, 72, 1019-1039. [CrossRef]
- Parodi-Rullan, R.M.; Javadov, S.; Fossati, S. Dissecting the Crosstalk between Endothelial Mitochondrial Damage, Vascular Inflammation, and Neurodegeneration in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Cells, 2021, 10, 2903. [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Parodi-Rullan, R.M.; Fossati, S.; Javadov, S. Beta-Amyloid Instigates Dysfunction of Mitochondria in Cardiac Cells. Cells, 2022, 11, 373. [CrossRef]
- Budd, S.L.; Nicholls, D.G. Mitochondria in the life and death of neurons. Essays Biochem., 1998, 33, 43-52. [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol Rev., 2000, 80, 315-360. [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum. Mol. Genet., 2009, 18, R169-R176. [CrossRef]
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci., 2009, 29, 9090-9103. [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol., 2018, 25, 59-70. [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers., 2021, 7, 33. [CrossRef]
- Nicholls,D.G., Ferguson,S.J. (2013) Bioenergetics 4. Academic Press, London.
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta, 2008, 1778, 1978-2021. [CrossRef]
- Kunji, E.R.; Aleksandrova, A.; King, M.S.; Majd, H.; Ashton, V.L.; Cerson, E.; Springett, R.; Kibalchenko, M.; Tavoulari, S.; Crichton, P.G. et al. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta, 2016, 1863, 2379-2393. [CrossRef]
- Gauba, E.; Chen, H.; Guo, L.; Du, H. Cyclophilin D deficiency attenuates mitochondrial F1Fo ATP synthase dysfunction via OSCP in Alzheimer’s disease. Neurobiol. Dis., 2019, 121, 138-147. [CrossRef]
- Beck, S.J.; Guo, L.; Phensy, A.; Tian, J.; Wang, L.; Tandon, N.; Gauba, E.; Lu, L.; Pascual, J.M.; Kroener, S. et al. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun., 2016, 7, 11483. [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de, B.L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta, 2008, 1777, 1289-1300. [CrossRef]
- Bobba, A.; Amadoro, G.; Petragallo, V.A.; Calissano, P.; Atlante, A. Dissecting the molecular mechanism by which NH2htau and Abeta1-42 peptides impair mitochondrial ANT-1 in Alzheimer disease. Biochim. Biophys. Acta, 2013, 1827, 848-860. [CrossRef]
- Atlante, A.; Valenti, D.; Latina, V.; Amadoro, G. Dysfunction of Mitochondria in Alzheimer’s Disease: ANT and VDAC Interact with Toxic Proteins and Aid to Determine the Fate of Brain Cells. Int. J. Mol. Sci., 2022, 23, 7722. [CrossRef]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature, 1995, 376, 509-514. [CrossRef]
- Khanna, R.; Wilson, S.M.; Brittain, J.M.; Weimer, J.; Sultana, R.; Butterfield, A.; Hensley, K. Opening Pandora’s jar: a primer on the putative roles of CRMP2 in a panoply of neurodegenerative, sensory and motor neuron, and central disorders. Future. Neurol., 2012, 7, 749-771.
- Hensley, K.; Venkova, K.; Christov, A.; Gunning, W.; Park, J. Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Mol. Neurobiol., 2011, 43, 180-191. [CrossRef]
- Hensley, K.; Kursula, P. Collapsin Response Mediator Protein-2 (CRMP2) is a Plausible Etiological Factor and Potential Therapeutic Target in Alzheimer’s Disease: Comparison and Contrast with Microtubule-Associated Protein Tau. J. Alzheimers. Dis., 2016, 53, 1-14. [CrossRef]
- Quach, T.T.; Moutal, A.; Khanna, R.; Deems, N.P.; Duchemin, A.M.; Barrientos, R.M. Collapsin Response Mediator Proteins: Novel Targets for Alzheimer’s Disease. J. Alzheimers. Dis., 2020, 77, 949-960. [CrossRef]
- Khanna, R.; Moutal, A.; Perez-Miller, S.; Chefdeville, A.; Boinon, L.; Patek, M. Druggability of CRMP2 for Neurodegenerative Diseases. ACS Chem. Neurosci., 2020, 11, 2492-2505. [CrossRef]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimers. Dis., 2006, 9, 309-317. [CrossRef]
- Cai, Z.; Zhao, Y.; Zhao, B. Roles of glycogen synthase kinase 3 in Alzheimer’s disease. Curr. Alzheimer Res., 2012, 9, 864-879. [CrossRef]
- Tsai, L.H.; Lee, M.S.; Cruz, J. Cdk5, a therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta, 2004, 1697, 137-142.
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res., 2012, 43, 655-662. [CrossRef]
- Gu, Y.; Hamajima, N.; Ihara, Y. Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry, 2000, 39, 4267-4275. [CrossRef]
- Cole, A.R.; Knebel, A.; Morrice, N.A.; Robertson, L.A.; Irving, A.J.; Connolly, C.N.; Sutherland, C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J. Biol. Chem., 2004, 279, 50176-50180. [CrossRef]
- Cole, A.R.; Noble, W.; van, A.L.; Plattner, F.; Meimaridou, R.; Hogan, D.; Taylor, M.; LaFrancois, J.; Gunn-Moore, F.; Verkhratsky, A. et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem., 2007, 103, 1132-1144. [CrossRef]
- Soutar, M.P.; Thornhill, P.; Cole, A.R.; Sutherland, C. Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Curr. Alzheimer Res., 2009, 6, 269-278. [CrossRef]
- Mokhtar, S.H.; Kim, M.J.; Magee, K.A.; Aui, P.M.; Thomas, S.; Bakhuraysah, M.M.; Alrehaili, A.A.; Lee, J.Y.; Steer, D.L.; Kenny, R. et al. Amyloid-beta-dependent phosphorylation of collapsin response mediator protein-2 dissociates kinesin in Alzheimer’s disease. Neural Regen. Res., 2018, 13, 1066-1080. [CrossRef]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain, 2008, 131, 90-108. [CrossRef]
- Rembutsu, M.; Soutar, M.P.; van, A.L.; Gourlay, R.; Hastie, C.J.; McLauchlan, H.; Morrice, N.A.; Cole, A.R.; Sutherland, C. Novel procedure to investigate the effect of phosphorylation on protein complex formation in vitro and in cells. Biochemistry, 2008, 47, 2153-2161. [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 Is Involved in Regulation of Mitochondrial Morphology and Motility in Neurons. Cells, 2021, 10, 2781. [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. Involvement of CRMP2 in Regulation of Mitochondrial Morphology and Motility in Huntington’s Disease. Cells, 2021, 10, 3172. [CrossRef]
- Xia, D.; Lianoglou, S.; Sandmann, T.; Calvert, M.; Suh, J.H.; Thomsen, E.; Dugas, J.; Pizzo, M.E.; DeVos, S.L.; Earr, T.K. et al. Novel App knock-in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol. Neurodegener., 2022, 17, 41. [CrossRef]
- Dubinsky, J.M. Intracellular calcium levels during the period of delayed excitotoxicity. J. Neurosci., 1993, 13, 623-631. [CrossRef]
- Brustovetsky,N., Brustovetsky,T. (2017) In Strack,S., Usachev,Y. (eds.), Techniques to investigate mitochondrial fucntion in neurons. New York, pp. 199-210.
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. The effect of mitochondrial calcium uniporter and cyclophilin D knockout on resistance of brain mitochondria to Ca(2+)-induced damage. J. Biol. Chem., 2021, 296, 100669. [CrossRef]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na(+)/Ca(2+) exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res., 2010, 88, 1317-1328.
- Brittain, J.M.; Chen, L.; Wilson, S.M.; Brustovetsky, T.; Gao, X.; Ashpole, N.M.; Molosh, A.I.; You, H.; Hudmon, A.; Shekhar, A. et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem., 2011, 286, 37778-37792. [CrossRef]
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R. et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death. Differ., 2018, 25, 542-572. [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Sridhar, A.; Pan, Y.; Cummins, T.R.; Meyer, J.S.; Brustovetsky, N. Energy Metabolism and Mitochondrial Superoxide Anion Production in Pre-symptomatic Striatal Neurons Derived from Human-Induced Pluripotent Stem Cells Expressing Mutant Huntingtin. Mol. Neurobiol., 2020, 57, 668-684. [CrossRef]
- Polster, B.M.; Nicholls, D.G.; Ge, S.X.; Roelofs, B.A. Use of potentiometric fluorophores in the measurement of mitochondrial reactive oxygen species. Methods Enzymol., 2014, 547, 225-250. [CrossRef]
- Kamo, N.; Muratsugu, M.; Hongoh, R.; Kobatake, Y. Membrane potential of mitochondria measured with an electrode sensitive to tetraphenyl phosphonium and relationship between proton electrochemical potential and phosphorylation potential in steady state. J. Membr. Biol., 1979, 49, 105-121. [CrossRef]
- Gawaz, M.; Douglas, M.G.; Klingenberg, M. Structure-function studies of adenine nucleotide transport in mitochondria. II. Biochemical analysis of distinct AAC1 and AAC2 proteins in yeast. J. Biol. Chem., 1990, 265, 14202-14208. [CrossRef]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem., 1994, 269, 27329-27336. [CrossRef]
- Brittain, J.M.; Piekarz, A.D.; Wang, Y.; Kondo, T.; Cummins, T.R.; Khanna, R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem., 2009, 284, 31375-31390. [CrossRef]
- Dustrude, E.T.; Perez-Miller, S.; Francois-Moutal, L.; Moutal, A.; Khanna, M.; Khanna, R. A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin. ), 2017, 11, 316-328. [CrossRef]
- Passarella, S.; Ostuni, A.; Atlante, A.; Quagliariello, E. Increase in the ADP/ATP exchange in rat liver mitochondria irradiated in vitro by helium-neon laser. Biochem. Biophys. Res. Commun., 1988, 156, 978-986. [CrossRef]
- Lustorff, J.; Schlimme, E. Does an inhibitor of mitochondrial adenylate kinase also affect oxidative phosphorylation? Experientia, 1976, 32, 298-299. [CrossRef]
- Klingenberg, M.; Grebe, K.; Scherer, B. The binding of atractylate and carboxy-atractylate to mitochondria. Eur. J. Biochem., 1975, 52, 351-363. [CrossRef]
- Klingenberg, M.; Buchholz, M. On the mechanism of bongkrekate effect on the mitochondrial adenine-nucleotide carrier as studied through the binding of ADP. Eur. J. Biochem., 1973, 38, 346-358. [CrossRef]
- Brustovetsky, T.; Khanna, R.; Brustovetsky, N. CRMP2 participates in regulating mitochondrial morphology and motility in Alzheimer’s disease. Cells, 2023, 12, 1287. [CrossRef]
- Moutal, A.; Francois-Moutal, L.; Perez-Miller, S.; Cottier, K.; Chew, L.A.; Yeon, S.K.; Dai, J.; Park, K.D.; Khanna, M.; Khanna, R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol. Neurobiol., 2015, 53, 1959-1976. [CrossRef]
- Moutal, A.; Chew, L.A.; Yang, X.; Wang, Y.; Yeon, S.K.; Telemi, E.; Meroueh, S.; Park, K.D.; Shrinivasan, R.; Gilbraith, K.B. et al. (S)-lacosamide inhibition of CRMP2 phosphorylation reduces postoperative and neuropathic pain behaviors through distinct classes of sensory neurons identified by constellation pharmacology. Pain, 2016, 157, 1448-1463. [CrossRef]
- Wang, S.; Ichinomiya, T.; Savchenko, P.; Devulapalli, S.; Wang, D.; Beltz, G.; Saito, T.; Saido, T.C.; Wagner, S.L.; Patel, H.H. et al. Age-Dependent Behavioral and Metabolic Assessment of App (NL-G-F/NL-G-F) Knock-in (KI) Mice. Front Mol. Neurosci., 2022, 15, 909989.
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett., 1997, 416, 15-18. [CrossRef]
- Vercesi, A.E.; Kowaltowski, A.J.; Grijalba, M.T.; Meinicke, A.R.; Castilho, R.F. The role of reactive oxygen species in mitochondrial permeability transition. Biosci. Rep., 1997, 17, 43-52. [CrossRef]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell, 2017, 16, 943-955. [CrossRef]
- Kent, A.C.; El Baradie, K.B.Y.; Hamrick, M.W. Targeting the Mitochondrial Permeability Transition Pore to Prevent Age-Associated Cell Damage and Neurodegeneration. Oxid. Med. Cell Longev., 2021, 2021, 6626484. [CrossRef]
- Morinaka, A.; Yamada, M.; Itofusa, R.; Funato, Y.; Yoshimura, Y.; Nakamura, F.; Yoshimura, T.; Kaibuchi, K.; Goshima, Y.; Hoshino, M. et al. Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal., 2011, 4, ra26. [CrossRef]
- Bertholet, A.M.; Natale, A.M.; Bisignano, P.; Suzuki, J.; Fedorenko, A.; Hamilton, J.; Brustovetsky, T.; Kazak, L.; Garrity, R.; Chouchani, E.T. et al. Mitochondrial uncouplers induce proton leak by activating AAC and UCP1. Nature, 2022, 606, 180-187. [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell, 2005, 120, 137-149. [CrossRef]
- Uchida, Y.; Ohshima, T.; Sasaki, Y.; Suzuki, H.; Yanai, S.; Yamashita, N.; Nakamura, F.; Takei, K.; Ihara, Y.; Mikoshiba, K. et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells, 2005, 10, 165-179. [CrossRef]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry, 2006, 45, 3134-3145. [CrossRef]
- Toba, J.; Nikkuni, M.; Ishizeki, M.; Yoshii, A.; Watamura, N.; Inoue, T.; Ohshima, T. PPARgamma agonist pioglitazone improves cerebellar dysfunction at pre-Abeta deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem. Biophys. Res. Commun., 2016, 473, 1039-1044. [CrossRef]
- Yang, Z.; Kuboyama, T.; Tohda, C. A Systematic Strategy for Discovering a Therapeutic Drug for Alzheimer’s Disease and Its Target Molecule. Front Pharmacol., 2017, 8, 340. [CrossRef]
- Chang, K.L.; Wong, L.R.; Pee, H.N.; Yang, S.; Ho, P.C. Reverting Metabolic Dysfunction in Cortex and Cerebellum of APP/PS1 Mice, a Model for Alzheimer’s Disease by Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Agonist. Mol. Neurobiol., 2019, 56, 7267-7283. [CrossRef]
- Isono, T.; Yamashita, N.; Obara, M.; Araki, T.; Nakamura, F.; Kamiya, Y.; Alkam, T.; Nitta, A.; Nabeshima, T.; Mikoshiba, K. et al. Amyloid-beta(2)(5)(-)(3)(5) induces impairment of cognitive function and long-term potentiation through phosphorylation of collapsin response mediator protein 2. Neurosci. Res., 2013, 77, 180-185. [CrossRef]
- Yamashita, N.; Ohshima, T.; Nakamura, F.; Kolattukudy, P.; Honnorat, J.; Mikoshiba, K.; Goshima, Y. Phosphorylation of CRMP2 (collapsin response mediator protein 2) is involved in proper dendritic field organization. J. Neurosci., 2012, 32, 1360-1365. [CrossRef]
- Belzacq, A.S.; Vieira, H.L.; Kroemer, G.; Brenner, C. The adenine nucleotide translocator in apoptosis. Biochimie, 2002, 84, 167-176. [CrossRef]
- Held, P. Determination of NADH concentrations with the SynergyTM 2 multi-detection microplate reader using fluorescence or absorbance. Agilent Application Note, 2021, 1-6.
- Brustovetsky, T.; Brittain, M.K.; Sheets, P.L.; Cummins, T.R.; Pinelis, V.; Brustovetsky, N. KB-R7943, an inhibitor of the reverse Na+ /Ca2+ exchanger, blocks N-methyl-D-aspartate receptor and inhibits mitochondrial complex I. Br. J. Pharmacol., 2011, 162, 255-270. [CrossRef]
- Hamilton, J.; Pellman, J.J.; Brustovetsky, T.; Harris, R.A.; Brustovetsky, N. Oxidative metabolism in YAC128 mouse model of Huntington’s disease. Hum. Mol. Genet., 2015, 24, 4862-4878. [CrossRef]
- Nicholls, D.G. Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods Mol. Biol., 2012, 810, 119-133.
- Wojtala, A.; Bonora, M.; Malinska, D.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Methods to monitor ROS production by fluorescence microscopy and fluorometry. Methods Enzymol., 2014, 542, 243-262.
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem., 2018, 293, 15652-15663. [CrossRef]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem., 2002, 80, 207-218. [CrossRef]
Figure 1.
CRMP2 is hyperphosphorylated at Thr 509/514 and Ser 522, but total CRMP2 is unchanged in lysates of brain cortices of 4-month-old APP-SAA KI mice (AD) compared to brain cortices lysates from B6J hAbeta mice (Control, Ctrl). (S)-LCM decreased CRMP2 phosphorylation. Lanes 1-3, Ctrl mice; lanes 4-6, AD mice treated with a vehicle; lanes 7-9, AD mice treated with (S)-LCM. (S)-LCM (10 mg/kg body weight) and a vehicle (Veh, 10 μl DMSO in 0.2 ml saline) were delivered by oral gavage for 7 days prior to analysis. Data are mean ± SD, N=3 biological replicates, **p<0.01, ***p<0.001.
Figure 1.
CRMP2 is hyperphosphorylated at Thr 509/514 and Ser 522, but total CRMP2 is unchanged in lysates of brain cortices of 4-month-old APP-SAA KI mice (AD) compared to brain cortices lysates from B6J hAbeta mice (Control, Ctrl). (S)-LCM decreased CRMP2 phosphorylation. Lanes 1-3, Ctrl mice; lanes 4-6, AD mice treated with a vehicle; lanes 7-9, AD mice treated with (S)-LCM. (S)-LCM (10 mg/kg body weight) and a vehicle (Veh, 10 μl DMSO in 0.2 ml saline) were delivered by oral gavage for 7 days prior to analysis. Data are mean ± SD, N=3 biological replicates, **p<0.01, ***p<0.001.
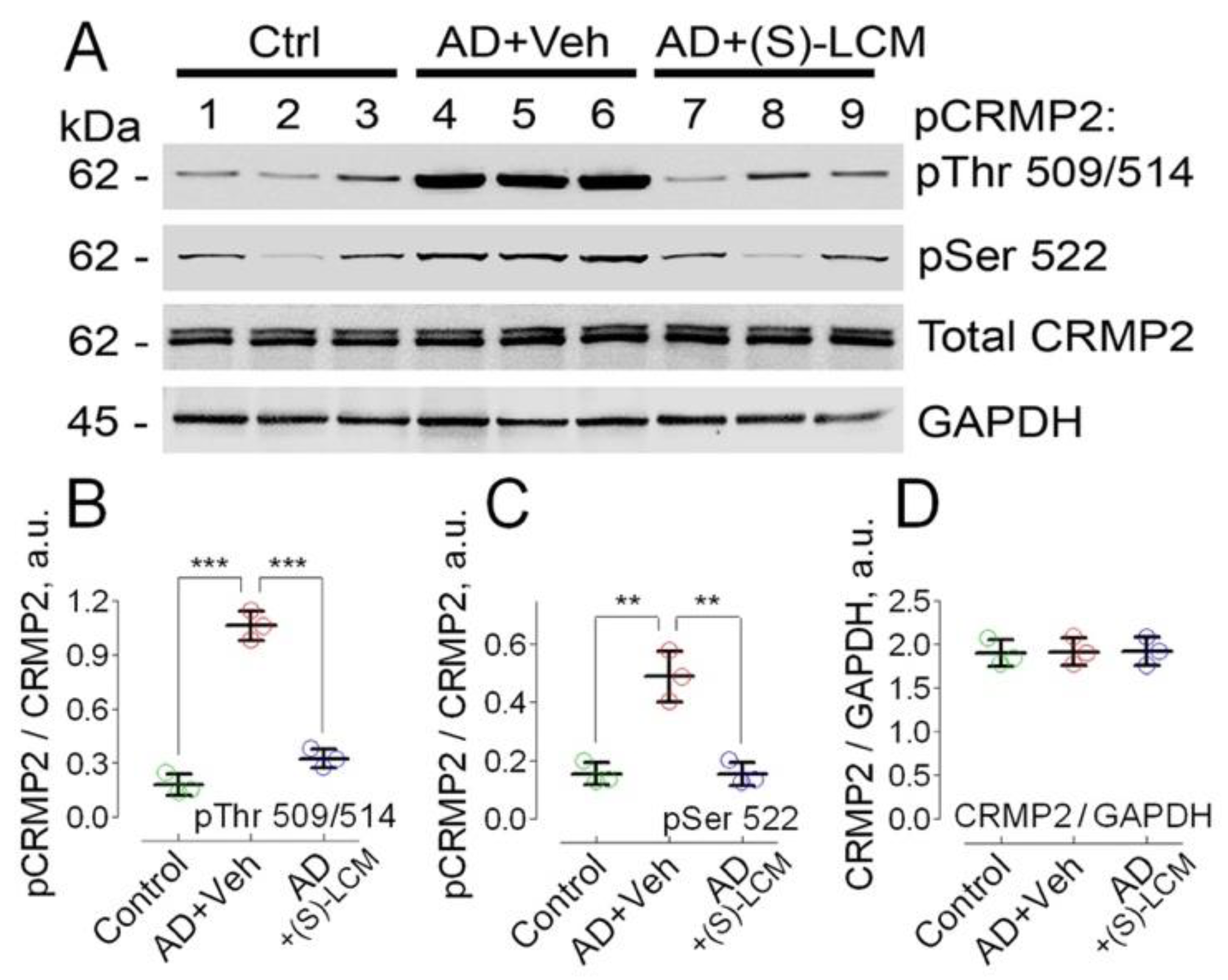
Figure 2.
CRMP2 is hyperphosphorylated at Tyr 32, Thr 509/514, Ser 522, and Thr 555 in cultured cortical neurons from APP-SAA KI mice (AD) compared to neurons from B6J hAbeta mice (Control, Ctrl). (S)-LCM prevented CRMP2 hyperphosphorylation at Thr 509/514 and Ser 522, but not at Tyr 32 and Thr 555. Cortical neurons were isolated from P1 AD and Ctrl mice of both sexes and cultured for 12-14 days in vitro (12-14 DIV). In A, representative immunoblots. B-E, statistical summaries based on densitometry data. Where indicated, neurons were treated with either 10 µM (S)-LCM or a vehicle (Veh, 0.01% DMSO) for the last 7 days prior to analysis. GAPDH is a loading control. Data are mean ± SD, N=4 experiments with cells from different platings, ***p<0.001.
Figure 2.
CRMP2 is hyperphosphorylated at Tyr 32, Thr 509/514, Ser 522, and Thr 555 in cultured cortical neurons from APP-SAA KI mice (AD) compared to neurons from B6J hAbeta mice (Control, Ctrl). (S)-LCM prevented CRMP2 hyperphosphorylation at Thr 509/514 and Ser 522, but not at Tyr 32 and Thr 555. Cortical neurons were isolated from P1 AD and Ctrl mice of both sexes and cultured for 12-14 days in vitro (12-14 DIV). In A, representative immunoblots. B-E, statistical summaries based on densitometry data. Where indicated, neurons were treated with either 10 µM (S)-LCM or a vehicle (Veh, 0.01% DMSO) for the last 7 days prior to analysis. GAPDH is a loading control. Data are mean ± SD, N=4 experiments with cells from different platings, ***p<0.001.
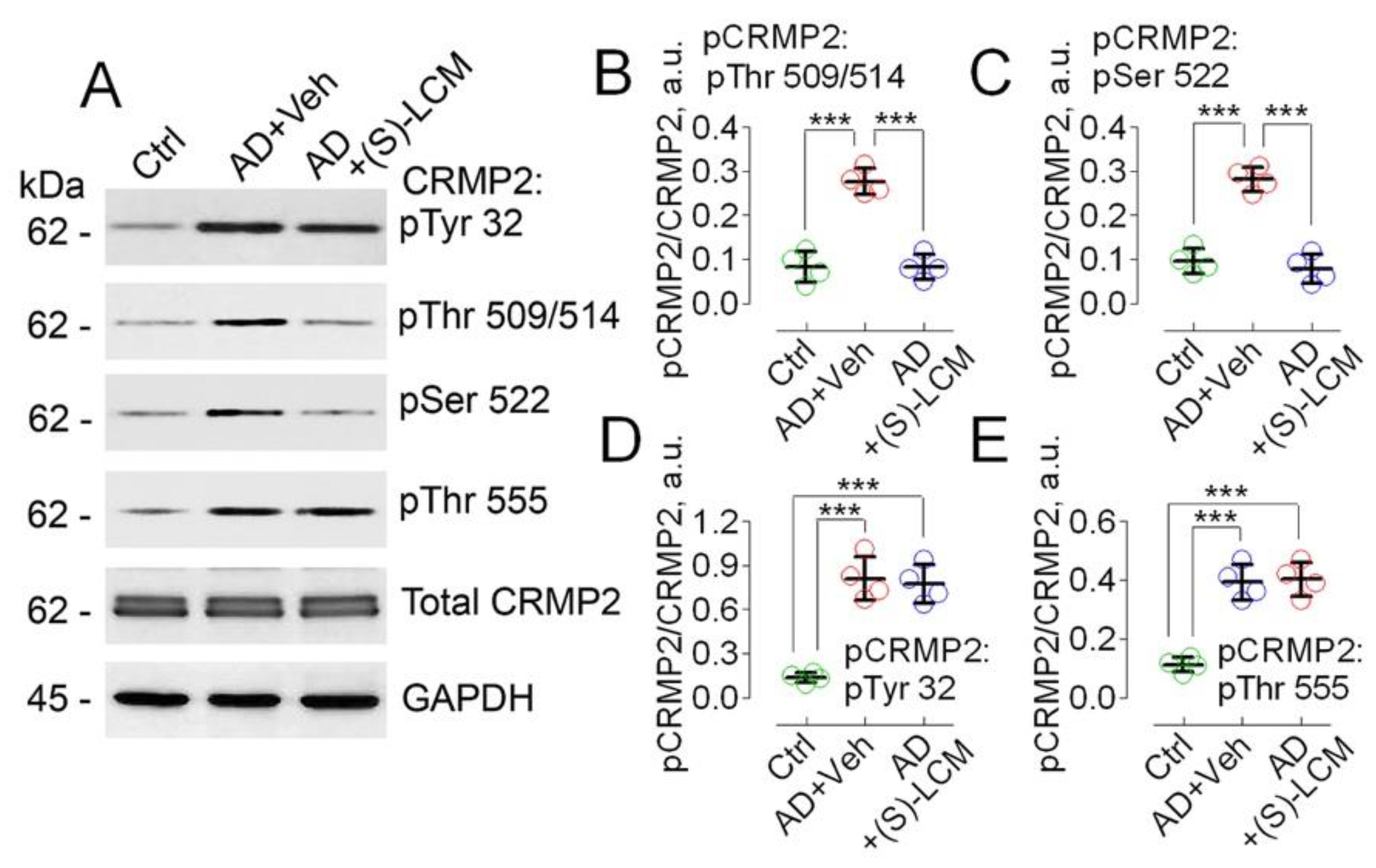
Figure 3.
Oxygen Consumption Rate (OCR) of cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice assessed with Seahorse XFe24 Analyzer, showing (S)-LCM improved respiration in AD neurons. The experiments were performed as we described earlier [57,88,89]. Oligomycin (1 μM, Oligo), inhibits F1F0-ATP synthase and decreases respiration associated with ATP regeneration; 2,4-dinitorphenol (60 μM, 2,4-DNP) uncouples OXPHOS and induces maximal respiration; rotenone (1 μM, Rot) and antimycin A (1 μM, Ant) are inhibitors of Complexes I and III, respectively, completely inhibit mitochondrial respiration. Where indicated, cells were treated with either a vehicle (Veh, 0.01% DMSO) or 10 µM (S)-LCM for 7 days prior to analysis. Cortical neurons were isolated from P1 AD and Ctrl mice of both sexes and cultured for 14 days in vitro (14 DIV). Data are mean ± SD, N=5 experiments with neurons from different platings.
Figure 3.
Oxygen Consumption Rate (OCR) of cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice assessed with Seahorse XFe24 Analyzer, showing (S)-LCM improved respiration in AD neurons. The experiments were performed as we described earlier [57,88,89]. Oligomycin (1 μM, Oligo), inhibits F1F0-ATP synthase and decreases respiration associated with ATP regeneration; 2,4-dinitorphenol (60 μM, 2,4-DNP) uncouples OXPHOS and induces maximal respiration; rotenone (1 μM, Rot) and antimycin A (1 μM, Ant) are inhibitors of Complexes I and III, respectively, completely inhibit mitochondrial respiration. Where indicated, cells were treated with either a vehicle (Veh, 0.01% DMSO) or 10 µM (S)-LCM for 7 days prior to analysis. Cortical neurons were isolated from P1 AD and Ctrl mice of both sexes and cultured for 14 days in vitro (14 DIV). Data are mean ± SD, N=5 experiments with neurons from different platings.
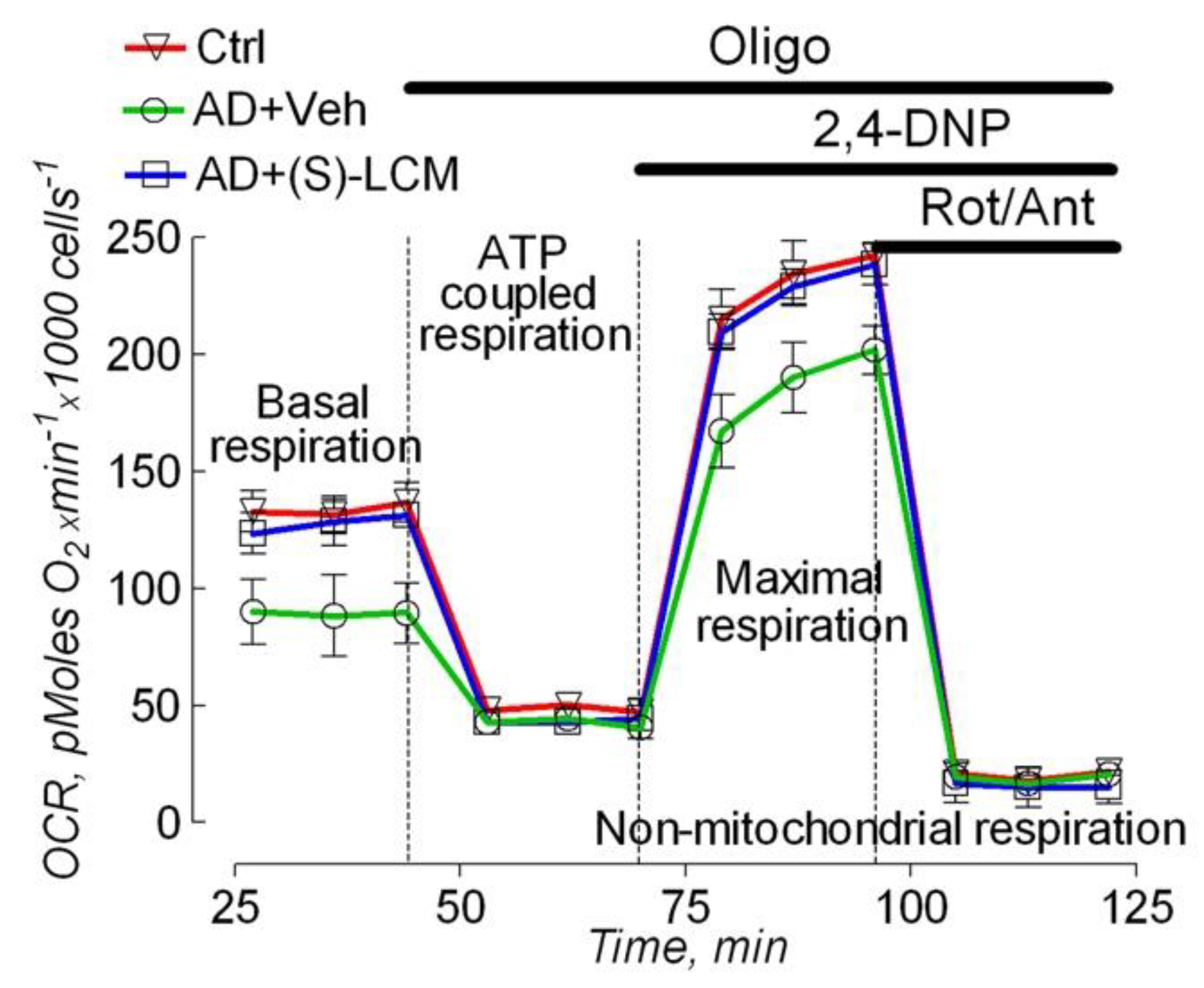
Figure 4.
Mitochondrial membrane potential in cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice, showing (S)-LCM prevented mitochondrial hyperpolarization in AD neurons. Neurons were cultured for 12-14 DIV and then co-stained with TMRM, a mitochondrial membrane potential probe, and NeuroFluor™ NeuO, neuronal marker. The mitochondrial membrane potential was assessed in individual cells by measuring TMRM signal using MetaMorph software [57,90]. The increased TMRM signal indicates higher mitochondrial membrane potential [90]. Bright field (Bf), TMRM, and NeuroFluor images of AD and Ctrl neurons are shown. Where indicated, cells were treated with either a vehicle (Veh, 0.01% DMSO) or 10 µM (S)-LCM for 7 days prior to analysis. In D, H, and L, mitochondria were depolarized with 1 μM FCCP as a positive control. In I, statistical summary of TMRM signals. Data are mean ± SD, ***p<0.001, N=96-102 cells from 5 different platings; a.u., arbitrary units. Scale bars, 10 μm.
Figure 4.
Mitochondrial membrane potential in cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice, showing (S)-LCM prevented mitochondrial hyperpolarization in AD neurons. Neurons were cultured for 12-14 DIV and then co-stained with TMRM, a mitochondrial membrane potential probe, and NeuroFluor™ NeuO, neuronal marker. The mitochondrial membrane potential was assessed in individual cells by measuring TMRM signal using MetaMorph software [57,90]. The increased TMRM signal indicates higher mitochondrial membrane potential [90]. Bright field (Bf), TMRM, and NeuroFluor images of AD and Ctrl neurons are shown. Where indicated, cells were treated with either a vehicle (Veh, 0.01% DMSO) or 10 µM (S)-LCM for 7 days prior to analysis. In D, H, and L, mitochondria were depolarized with 1 μM FCCP as a positive control. In I, statistical summary of TMRM signals. Data are mean ± SD, ***p<0.001, N=96-102 cells from 5 different platings; a.u., arbitrary units. Scale bars, 10 μm.
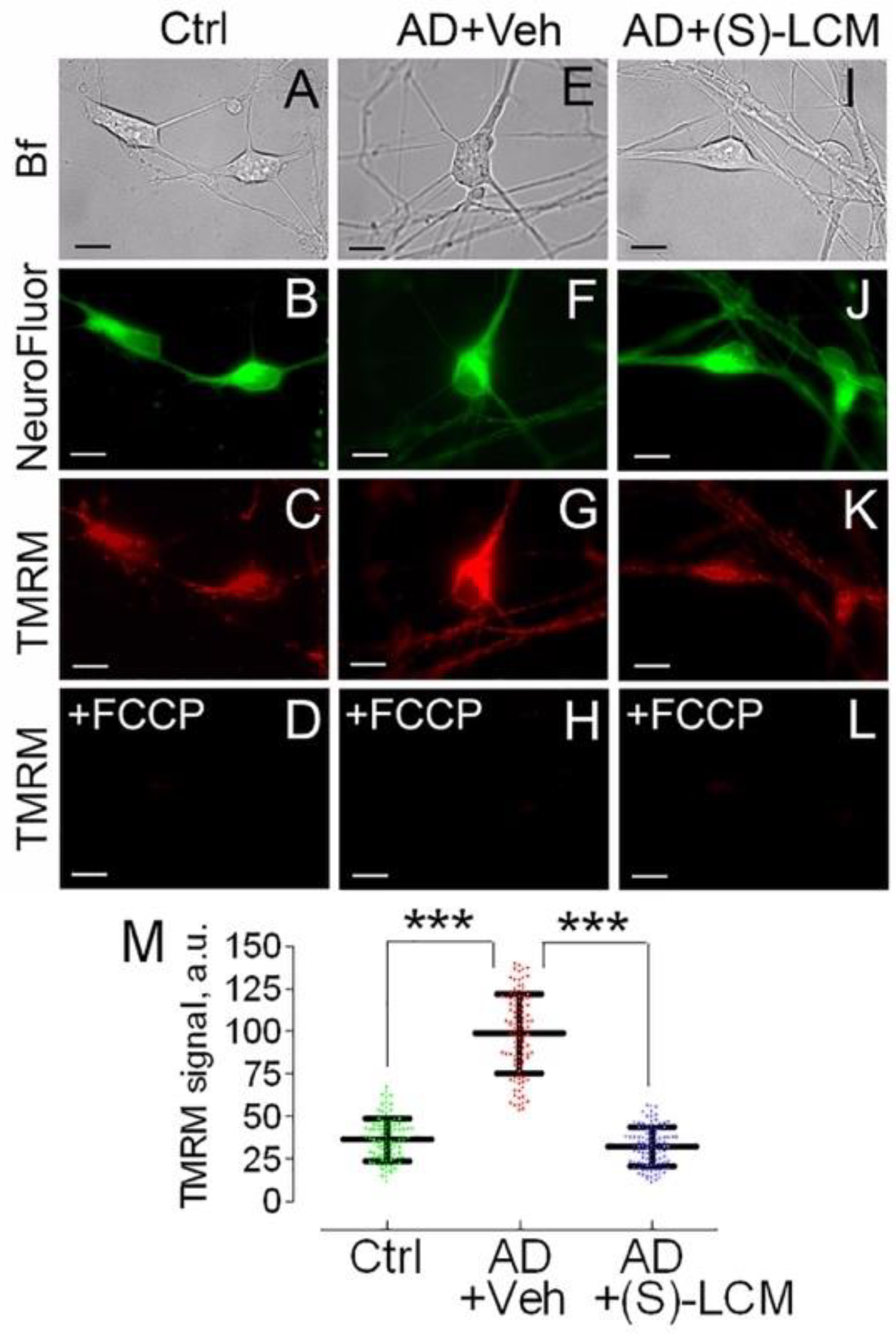
Figure 5.
Mitochondrial superoxide anion (O2-) production in cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice. (S)-LCM reduced O2- production in AD neurons. Neurons were cultured for 12-14 DIV and were detected with NeuroFluor™ NeuO, neuronal marker. O2- was evaluated by measuring MitoSOX Red fluorescence using MetaMorph software [57]. The increased MitoSOX signal indicates higher O2- level [91]. Bright field (Bf), MitoSOX, and NeuroFluor images of AD and Ctrl neurons are shown. Cells were treated with either a vehicle (Veh, 0.01% DMSO) (E-H) or 10 µM (S)-LCM (I-L) for 7 days prior to analysis. In A-C, E-G, and I-K cells were incubated in the bath solution for 8 minutes before taking images and then exposed to 10 μM Antimycin A (Ant) as a positive control for 10 minutes (D, H, L). In M, statistical summary of MitoSOX signals. Data are mean ± SD, ***p<0.001, N=95-100 cells from 5 different platings; a.u., arbitrary units. Scale bars, 10μm.
Figure 5.
Mitochondrial superoxide anion (O2-) production in cultured cortical neurons from APP-SAA KI (AD) and B6J hAbeta (Control, Ctrl) mice. (S)-LCM reduced O2- production in AD neurons. Neurons were cultured for 12-14 DIV and were detected with NeuroFluor™ NeuO, neuronal marker. O2- was evaluated by measuring MitoSOX Red fluorescence using MetaMorph software [57]. The increased MitoSOX signal indicates higher O2- level [91]. Bright field (Bf), MitoSOX, and NeuroFluor images of AD and Ctrl neurons are shown. Cells were treated with either a vehicle (Veh, 0.01% DMSO) (E-H) or 10 µM (S)-LCM (I-L) for 7 days prior to analysis. In A-C, E-G, and I-K cells were incubated in the bath solution for 8 minutes before taking images and then exposed to 10 μM Antimycin A (Ant) as a positive control for 10 minutes (D, H, L). In M, statistical summary of MitoSOX signals. Data are mean ± SD, ***p<0.001, N=95-100 cells from 5 different platings; a.u., arbitrary units. Scale bars, 10μm.
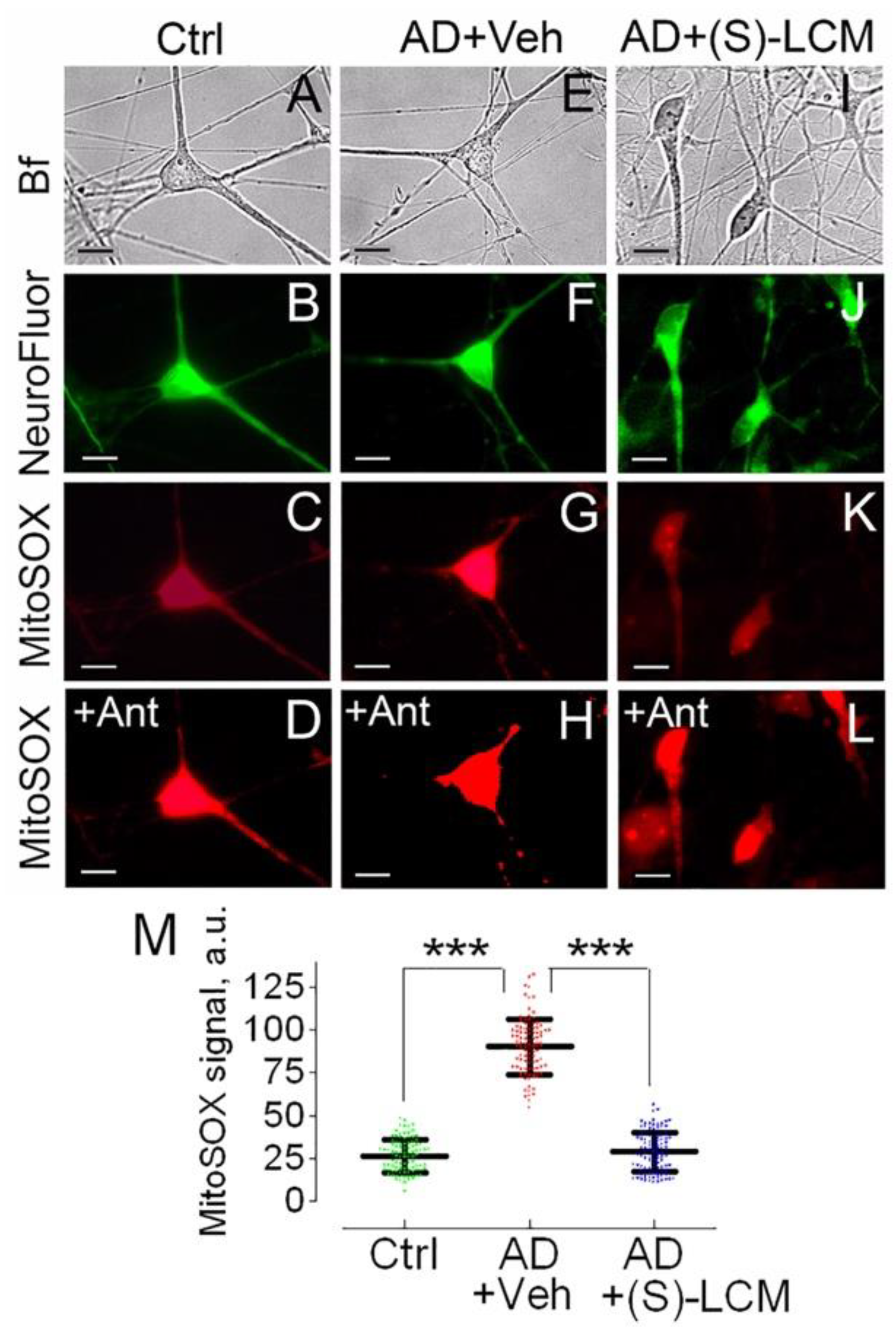
Figure 6.
Respiration (red traces) and membrane potential (black traces) of synaptic mitochondria isolated from cortices of 4-month-old APP-SAA KI (AD) and age-matched B6J hAbeta (Control, Ctrl) mice. (S)-LCM improved responses to ADP and 2,4-dinitrophenol (DNP) in AD mitochondria. Mitochondria were isolated from 4-month old AD and Ctrl mice of both sexes. AD mice were treated with either a vehicle (Veh, 10 µl of DMSO in 0.2 ml saline) or 10 mg (S)-LCM/kg body weight delivered by oral gavage for 7 days before analysis. In A-C, representative measurements of mitochondrial respiration and membrane potential. Mitochondria were incubated at 37°C in KCl-based medium with 1 mM malate plus 3 mM pyruvate [53,92]. Respiration was measured with a Clark-type oxygen electrode, mitochondrial membrane potential was followed with a tetraphenylphosphonium (TPP+)-sensitive electrode [53,92,93]. In D, statistical summary. V2, respiration rate before ADP addition; V3, respiration stimulated by 300 μM ADP; V4, respiration after ADP depletion; VDNP, respiration with 60 μM of 2,4-dinitrophenol (DNP). Data are mean ± SD, N=5 experiments, ***p<0.001.
Figure 6.
Respiration (red traces) and membrane potential (black traces) of synaptic mitochondria isolated from cortices of 4-month-old APP-SAA KI (AD) and age-matched B6J hAbeta (Control, Ctrl) mice. (S)-LCM improved responses to ADP and 2,4-dinitrophenol (DNP) in AD mitochondria. Mitochondria were isolated from 4-month old AD and Ctrl mice of both sexes. AD mice were treated with either a vehicle (Veh, 10 µl of DMSO in 0.2 ml saline) or 10 mg (S)-LCM/kg body weight delivered by oral gavage for 7 days before analysis. In A-C, representative measurements of mitochondrial respiration and membrane potential. Mitochondria were incubated at 37°C in KCl-based medium with 1 mM malate plus 3 mM pyruvate [53,92]. Respiration was measured with a Clark-type oxygen electrode, mitochondrial membrane potential was followed with a tetraphenylphosphonium (TPP+)-sensitive electrode [53,92,93]. In D, statistical summary. V2, respiration rate before ADP addition; V3, respiration stimulated by 300 μM ADP; V4, respiration after ADP depletion; VDNP, respiration with 60 μM of 2,4-dinitrophenol (DNP). Data are mean ± SD, N=5 experiments, ***p<0.001.
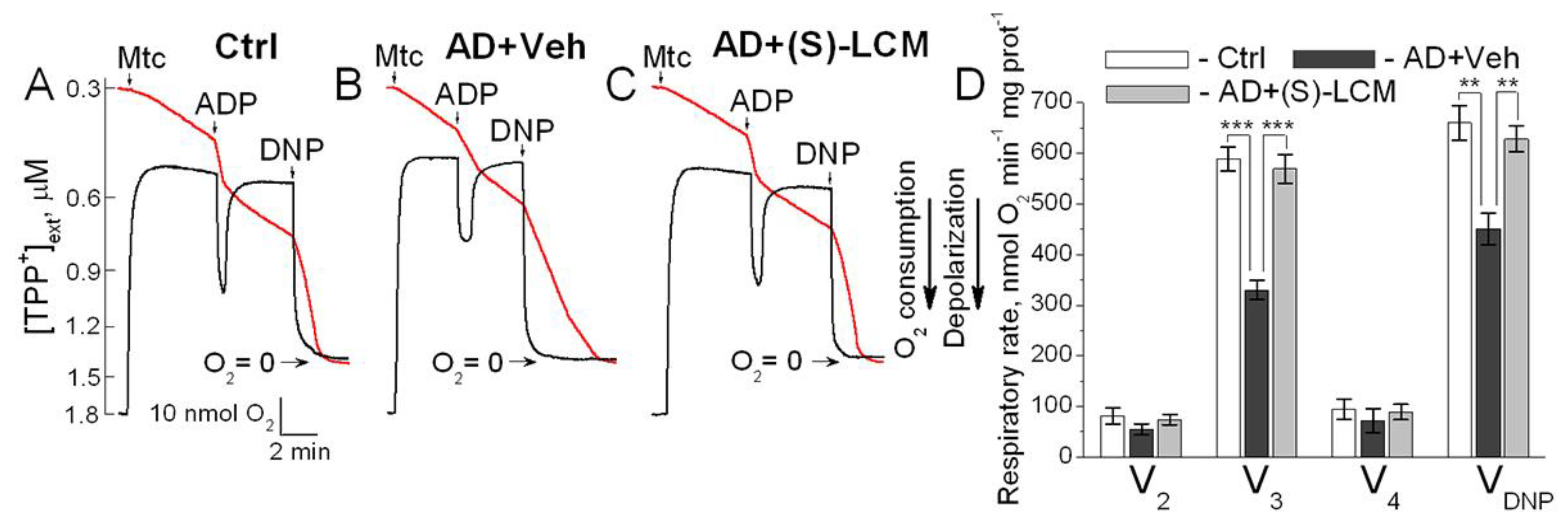
Figure 7.
CRMP2 co-immunoprecipitated with the adenine nucleotide translocase (ANT) in brain cortical synaptic mitochondria isolated from 4-month-old B6J hAbeta mice (Control, Ctrl). In mitochondria from 4-month-old APP-SAA KI mice (AD), CRMP2 dissociated from the ANT. (S)-LCM prevented CRMP2 dissociation from the ANT in AD mitochondria. In A and B, representative immunoblots with cortical synaptic mitochondria from AD and Ctrl mice. In A, immunoprecipitation was performed with anti-CRMP2 antibody, and the ANT was detected with anti-ANT1/2 antibody. In B, immunoprecipitation was performed with anti-ANT1/2 antibody, and CRMP2 was detected with anti-CRMP2 antibody. Where indicated, AD mice were treated with (S)-LCM (10 mg/kg body weight) or a vehicle (Veh, 10 μl DMSO in 0.2 ml saline), delivered by oral gavage for 7 days prior to analysis. Mitochondria were isolated from mice of both sexes. The Input was 5% of total protein used in pull-down procedure. Representative data from N=5 biological repeats are shown.
Figure 7.
CRMP2 co-immunoprecipitated with the adenine nucleotide translocase (ANT) in brain cortical synaptic mitochondria isolated from 4-month-old B6J hAbeta mice (Control, Ctrl). In mitochondria from 4-month-old APP-SAA KI mice (AD), CRMP2 dissociated from the ANT. (S)-LCM prevented CRMP2 dissociation from the ANT in AD mitochondria. In A and B, representative immunoblots with cortical synaptic mitochondria from AD and Ctrl mice. In A, immunoprecipitation was performed with anti-CRMP2 antibody, and the ANT was detected with anti-ANT1/2 antibody. In B, immunoprecipitation was performed with anti-ANT1/2 antibody, and CRMP2 was detected with anti-CRMP2 antibody. Where indicated, AD mice were treated with (S)-LCM (10 mg/kg body weight) or a vehicle (Veh, 10 μl DMSO in 0.2 ml saline), delivered by oral gavage for 7 days prior to analysis. Mitochondria were isolated from mice of both sexes. The Input was 5% of total protein used in pull-down procedure. Representative data from N=5 biological repeats are shown.
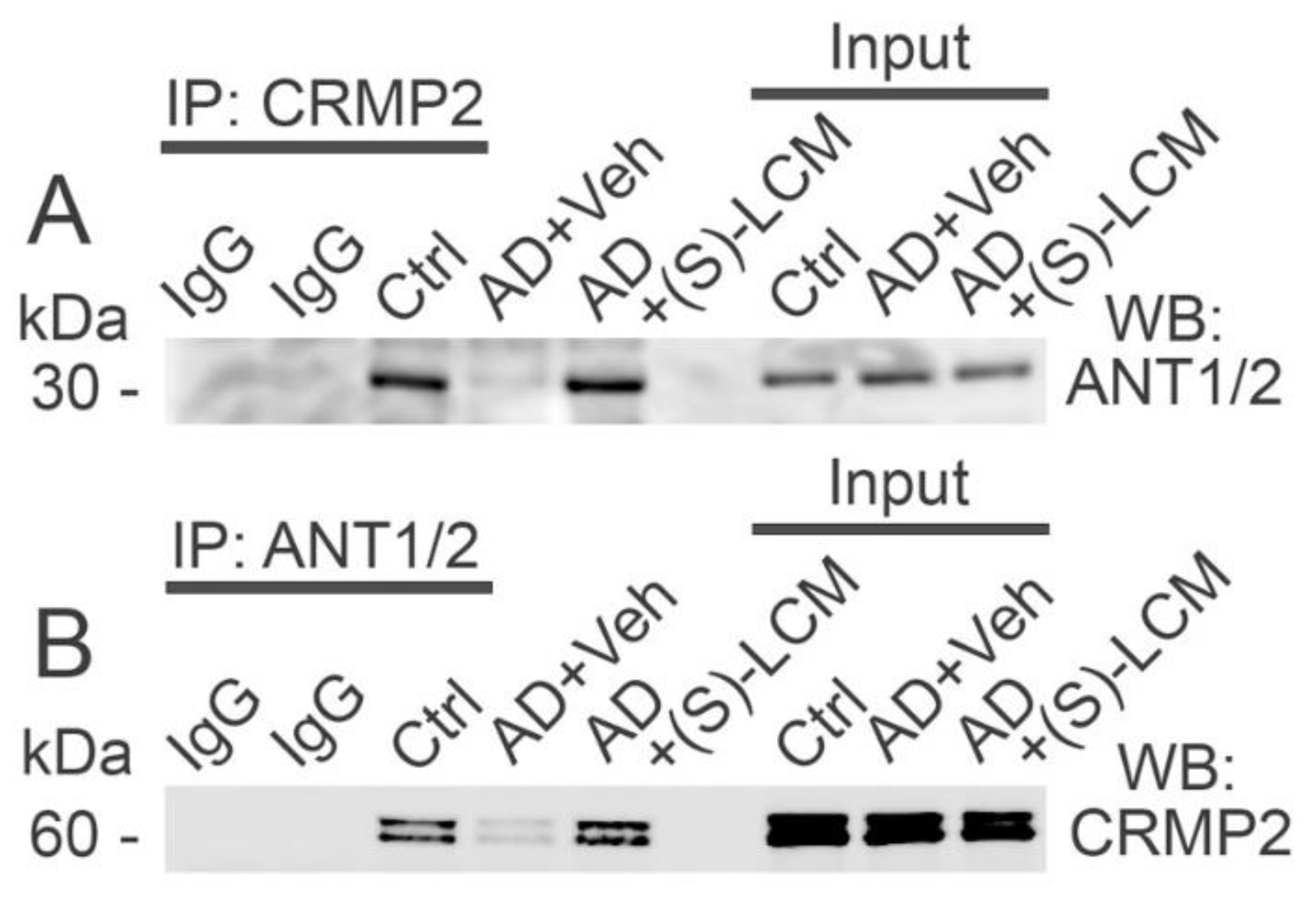
Figure 8.
The ANT activity is reduced in synaptic mitochondria isolated from brain cortices of 4-month-old APP-SAA KI (AD) compared with mitochondria from age-matched B6J hAbeta (Control, Ctrl) mice. Pretreatment of AD mice with (S)-LCM improved ANT activity. Where indicated, AD mice were treated with (S)-LCM (10 mg/kg body weight) or a vehicle (Veh, 10 μl DMSO in 0.2 ml saline), delivered by oral gavage for 7 days prior to analysis. As a control, carboxyatractyloside (CAT, 5 μM) and bongkrekic acid (BKA, 5 μM) completely inhibited the ANT. In A, representative fluorescence traces. In B, statistical summary of NADPH measurements. Data are mean ± SD, **p<0.01, ***p<0.001, N=5 separate experiments.
Figure 8.
The ANT activity is reduced in synaptic mitochondria isolated from brain cortices of 4-month-old APP-SAA KI (AD) compared with mitochondria from age-matched B6J hAbeta (Control, Ctrl) mice. Pretreatment of AD mice with (S)-LCM improved ANT activity. Where indicated, AD mice were treated with (S)-LCM (10 mg/kg body weight) or a vehicle (Veh, 10 μl DMSO in 0.2 ml saline), delivered by oral gavage for 7 days prior to analysis. As a control, carboxyatractyloside (CAT, 5 μM) and bongkrekic acid (BKA, 5 μM) completely inhibited the ANT. In A, representative fluorescence traces. In B, statistical summary of NADPH measurements. Data are mean ± SD, **p<0.01, ***p<0.001, N=5 separate experiments.
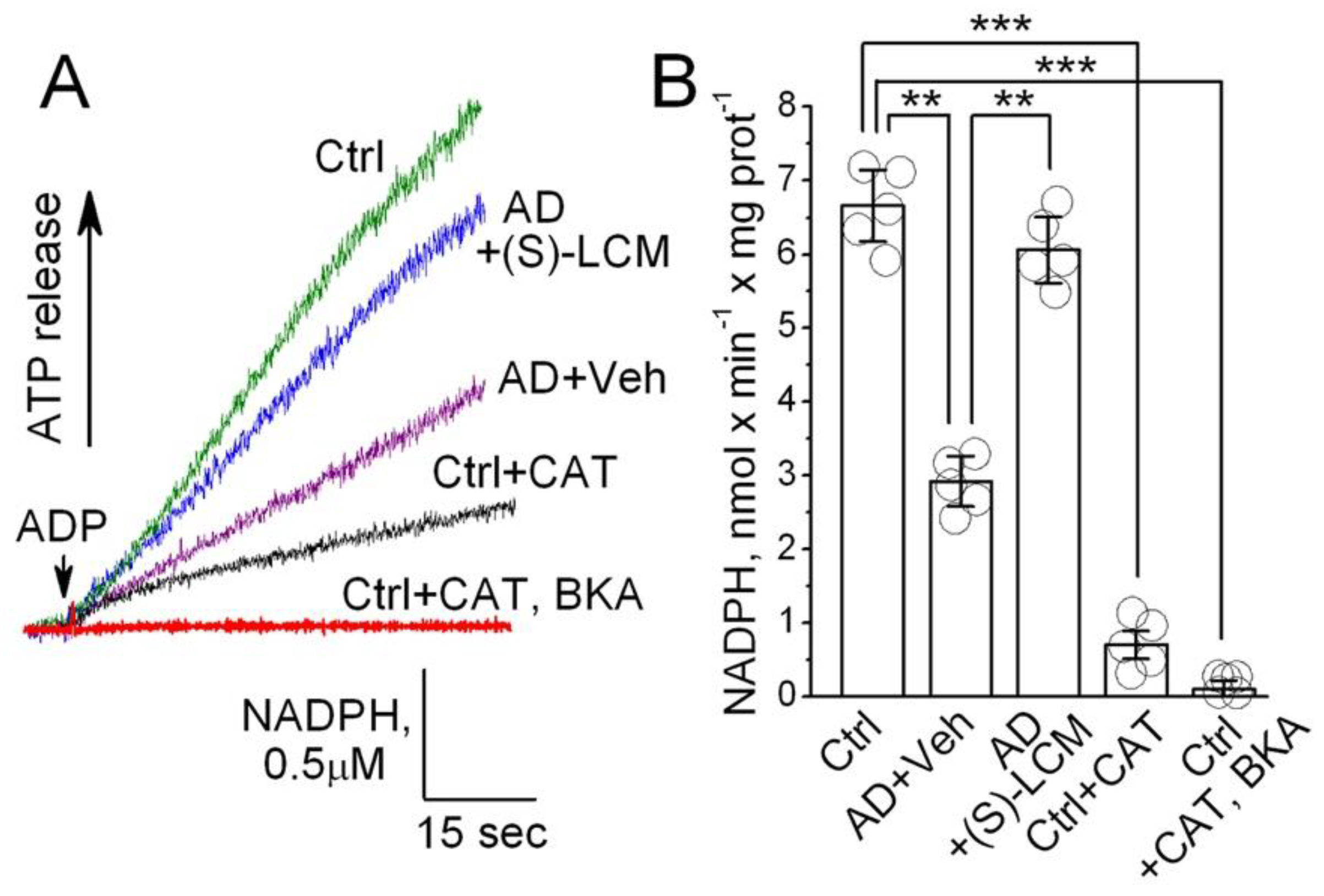
Figure 9.
The ANT activity is increased in the ANT proteoliposomes treated with recombinant CRMP2 (rCRMP2). In A, representative fluorescence traces. The ANT proteoliposomes were loaded with 20 mM ATP. Where indicated, recombinant CRMP2 (rCRMP2, 10 μg/ml) was present and then 100 μM ADP was added. The coupled ATP detecting system consisted of 2.5 mM glucose, 1.0 U/ml hexokinase, 1.0 U/ml glucose-6-phosphate dehydrogenase, 0.5 mM NADP+, and 10 μM P1,P5-Di(adenosine-5′)pentaphosphate, an inhibitor of adenylate kinase. As a control, carboxyatractyloside (CAT, 5 μM) and bongkrekic acid (BKA, 5 μM) completely inhibited the ANT. In D, statistical summary of NADPH measurements. Data are mean ± SD. **p<0.01, ***p<0.001, N=5 separate experiments.
Figure 9.
The ANT activity is increased in the ANT proteoliposomes treated with recombinant CRMP2 (rCRMP2). In A, representative fluorescence traces. The ANT proteoliposomes were loaded with 20 mM ATP. Where indicated, recombinant CRMP2 (rCRMP2, 10 μg/ml) was present and then 100 μM ADP was added. The coupled ATP detecting system consisted of 2.5 mM glucose, 1.0 U/ml hexokinase, 1.0 U/ml glucose-6-phosphate dehydrogenase, 0.5 mM NADP+, and 10 μM P1,P5-Di(adenosine-5′)pentaphosphate, an inhibitor of adenylate kinase. As a control, carboxyatractyloside (CAT, 5 μM) and bongkrekic acid (BKA, 5 μM) completely inhibited the ANT. In D, statistical summary of NADPH measurements. Data are mean ± SD. **p<0.01, ***p<0.001, N=5 separate experiments.
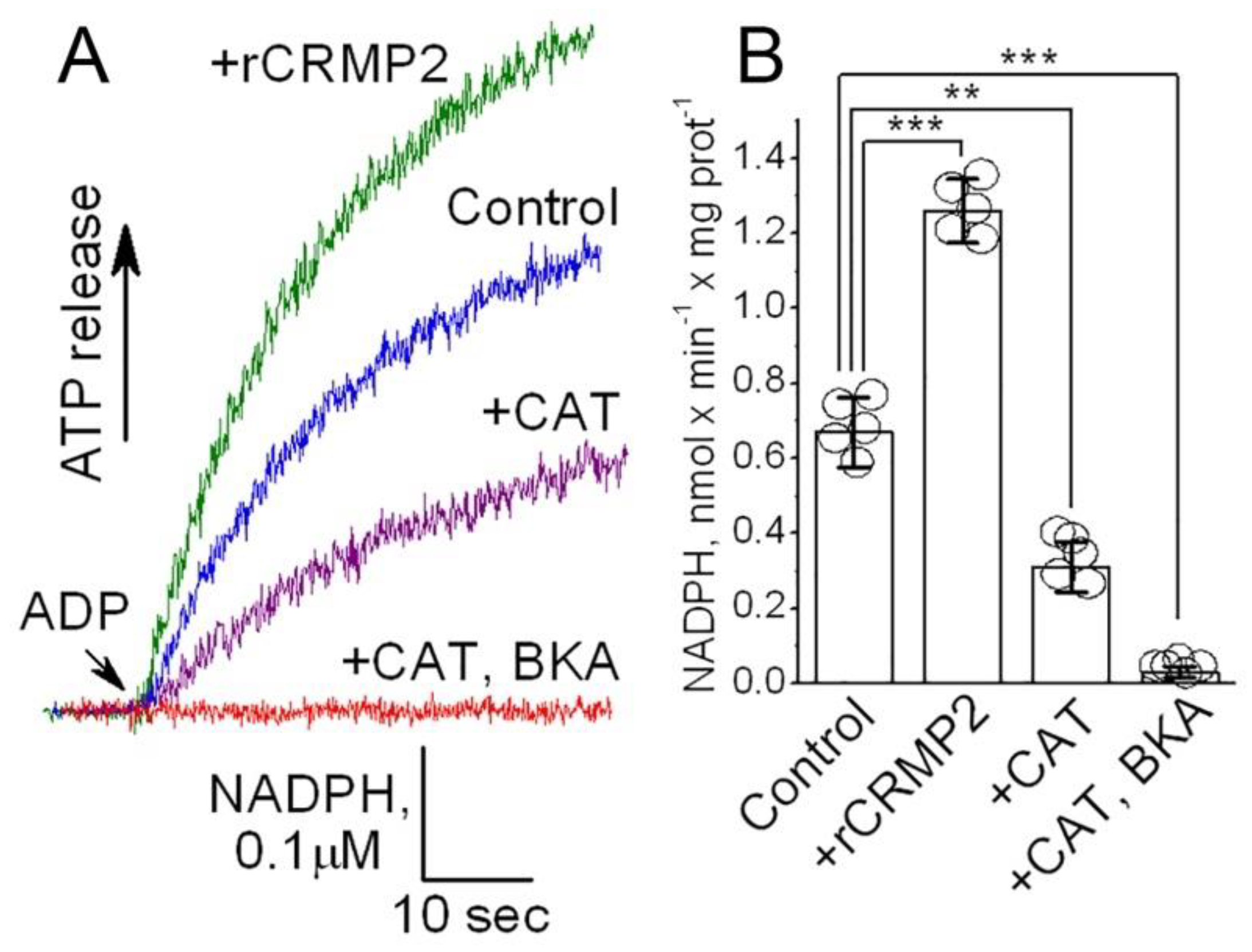
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



