
Submitted:
14 April 2025
Posted:
15 April 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The ultimate measurement of viral fitness is the ability to maintain high prevalence rates in its host species. Effective transmission, efficient replication, and rapid immune evasion all lead to this end. During the past five years, SARS-CoV-2 has successfully adapted to the human host and established human reservoirs for long term coexistence with mankind. We have observed innovative synergistic mutations in the Spike protein to improve receptor binding. Adaptation to cells of the upper respiratory tract has shortened the incubation period and facilitated viral spread. Such improvements allowed for immune escape mutations even though they may reduce replicative fitness. Adaptive mutations have resulted in intermittent selective sweeps by dominant variants. However, there are limitations to functional improvements. Receptor binding affinity of the Spike protein peaked in 2022-2023. Accumulation of fixed mutations plateaued after the advent of BA.2.86/JN.1 at the turn of 2023 and 2024. Purifying selection has been the main force working on nonsynonymous mutations in the Omicron group and overall fitness effects of missense mutations in major viral proteins have been declining. Moreover, because of weak selection on synonymous mutations, codon adaptation index in the human host has been decreasing among Omicron subvariants. Consequently, Omicron lineages replicated in cell cultures less efficiently than the original virus, and recent Omicron lineages showed signs of further attenuation in animal models. Viral attenuation in the human population manifested as declining COVID-19-related mortality even with high prevalence of infections.
Keywords:
SARS-CoV-2
; COVID-19
; Evolution
; Mutation
; Muller's ratchet
Introduction
In the study of real time evolutionary processes, no organism has been observed and documented as intensively and concertedly as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19. Millions of genomic sequences have been analyzed during the pandemic. With its short generation time, high mutation rate, large surplus population, and strong natural selection, variants of SARS-CoV-2 have showcased “speciation”, selective sweeps, and “extinction” in matters of weeks and months. The dazzling drama of alphabetical “dynasties” (Figure 1A and 1B) not only gave us insights about the evolutionary trajectory of a zoonotic RNA virus and its interaction with the human host but also provided us with an opportunity to learn general trends in molecular evolution which would take deep geological time to be appreciable in higher organisms.
SARS-CoV-2 is the third zoonotic coronavirus of the century. While severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) went extinct and Middle East respiratory syndrome coronavirus (MERS-CoV) is now only reported sporadically in Saudi Arabia, SARS-CoV-2 has reigned in every corner of the world and infections remain prevalent year-round. SARS-CoV-2 demonstrated that the ultimate measurement of viral fitness is the ability to maintain high prevalence rates in its host species. Effective human-to-human transmission, efficient replication in human cells, and rapid immune evasion are all means toward this end. On the molecular level, innovative mutations have enabled the Spike protein of SARS-CoV-2 to enhance binding affinity toward its primary receptor on human cells, angiotensin-converting enzyme 2 (ACE2). Perhaps related to this gain of molecular function, the virus increased its ability to infect cells of the upper airway in preference to the lungs because ACE2 is more concentrated in the nasal epithelium [1]. Tropism toward the nasal tissues facilitated viral shedding and probably contributed to the shortening of the incubation period and increased transmissibility. In addition to its role in receptor binding, the Spike protein is also the primary target of neutralizing antibodies. For these reasons, the Spike protein, especially its receptor-binding domain (RBD), has experienced higher rates of nonsynonymous mutations than any other protein of the virus.
However, functional improvements have theoretical and practical limits. Early in the pandemic, Zahradník et al demonstrated by in vitro evolution that there is an optimal configuration of the Spike protein (RBD-62) with an ACE2-binding affinity that is 1000-fold stronger than wild type [2]. Many variants of SARS-CoV-2 have employed the same mutations predicted by the in vitro experiment. Meanwhile, the virus must constantly change the Spike protein to evade neutralizing antibodies regardless of the effects of the mutations on receptor binding. In addition, evolution of the viral genome is not only driven by functional advantages, but also by the preferred directions of the host-initiated mutagenic mechanisms such as the RNA editing proteins APOBEC (Apolipoprotein B mRNA Editing Catalytic Polypeptide-like), ADAR (Adenosine Deaminase Acting on RNA), and ZAP (zinc finger antiviral proteins) [3,4,5]. Moreover, a significant proportion of mutations are not selectable and therefore subject to random drifting [6]. For these reasons, ideal conformations such as RBD-62, are probably not achievable in nature.
The purpose of this review is to address such questions as: Where is SARS-CoV-2 in its evolutionary trajectory after five years of circulation in humans? Do we expect more transmissible or more virulent variants in the future? Or is the virus losing replicative fitness and virulence irreversibly? Here we discuss several observations that indicate that genomic evolution of SARS-CoV-2 has reached a plateau and is probably going downhill.
1. Accumulation of Fixed Mutations Is Slowing Down
While the majority (over 60%) of the ~30,000 nucleotides of the SARS-CoV-2 genome have mutated at least once [4,7], only a small proportion of the mutations have been selected and fixed [8]. Most synonymous mutations are near neutral, while most nonsynonymous mutations are deleterious [6]. Most of the fixed mutations are in the Spike protein. By the beginning of 2025, SARS-CoV-2 had accumulated over 60 mutations in the 1273-residue-long Spike protein, 30 of which in the 223-residue-long RBD [9,10]. On the other hand, there are altogether about 100 fixed nonsynonymous mutations in the SARS-CoV-2 genome coding for 29 proteins totaling over 9000 amino acid residues [10]. In other words, the overall rate of fixed amino acid mutations is ~1% while there are ~5% fixed mutations in Spike and over 13% in the RBD.
1.1. Cooperative Emergence of Multiple Affinity-Enhancing Mutations Followed by Immune Escape Mutations
Buildup of missense mutations in the Spike has been gradual with two brief periods of acceleration, one at the emergence of Omicron BA.1, and the other at the emergence of Omicron BA.2.86 (Figure 1C) [10]. The sudden increase in missense mutations in BA.1 also manifested as an increase in the ratio between nonsynonymous mutations and synonymous mutations (dn/ds or ω, Figure 1D).
Although many missense mutations in the Spike protein experienced positive selection [8], only about a dozen of them enhanced receptor binding affinity [11,12,13,14,15]. Synergism and epistasis among affinity-enhancing mutations played a major role in the mutation count jumps, meaning certain mutations facilitated the fixation of other mutations, generating new modes of interactions between the RBD and ACE2 [2,17,26]. For example, the N501Y mutation turned the affinity-reducing Q498R mutation to an affinity-enhancing mutation. Synergism between N501Y and Q498R reconfigured the RBD-ACE2 interaction in the Omicron variant [18]. Synergistic affinity-enhancing mutations were often followed by immune escape mutations which provided more prominent selective advantages than affinity enhancement [12,19,20,21]. The affinity-enhancing mutations in BA.1 (S477N, Q493R, Q498R, N501Y) first appeared in the original Omicron variant (B.1.1.529) and the affinity-reducing mutations (including three deletions) were added subsequently for immune escape, enabling BA.1 to spread quickly, displacing the Delta variant in a few months (Figure 1A) [10,12].
Likewise, R403K did not increase ACE2-binding affinity in the earlier B.1.1 lineage but did in the BA.2 lineage background [22]. It first appeared in BA.2.86. Although BA2.86, with its high receptor-binding affinity, was recognized for its increase in frequency, it never rose to dominant status (with a peak frequency of ~9% in the US [23]). However, one more mutation, L455S, turned BA2.86 into JN.1, which quickly wiped out all other subvariants to dominate the variant landscape (Figure 1 A and B). L455S significantly reduced the ACE2-binding affinity of the RBD but provided JN.1 with strong immune-evading ability along with enhanced fusogenicity and improved cell entry [20,21,24].
Similarly, Q493E enhanced ACE2 binding only in the presence of L455S and F456L, found in some JN.1 sublineages. Q493E emerged independently in KP.3 and LP.8 [17]. Subsequently, a deletion, S31del, developed in KP.3.1 to produce KP.3.1.1. Although the deletion itself reduced the ability of the virus to infect cells by membrane fusion, it granted KP.3.1.1 a significant competitive edge over its parent by immune evasion [25,26]. S31del also independently emerged in other JN.1 sublineages such as KP.2.3 and LP.8, showing that S31 was a prominent immune target in the human population during the past year.
1.2. Convergence, Flip, and Reversion
Even though the Spike protein experienced strong positive selection [8], natural mutations in the Spike during the pandemic indicated that there are limited modes of receptor binding that the virus can evolve into. For this reason, mutations in the S gene tend to be convergent, recurrent, and cyclical. Table 1 lists some of the recurrent Spike mutations. Multiple variants resorted to the same tricks again and again.
The FLip mutants showed that the virus can swap positions of two adjacent amino acids in the Spike protein to evade neutralizing antibodies and to improve receptor binding. Multiple XBB and JN.1 sublineages swapped the positions of L455 and F456 via the FLip mutations (L455F + F456L) [16,27]. Both mutations, especially L455F, dampened receptor binding when acting alone [16], and F456L also reduced viral infectivity [15], which is probably why they did not develop early in the pandemic. However, more than three years later, F456L showed up, presumably because it enabled the virus to evade antibodies developed against earlier lineages. Subsequent L455F mutation effectively restored receptor binding affinity and further helped the virus to penetrate herd immunity [16].
RBD mutations also reverted depending on the genetic background. The Q493R mutation in BA.1 and BA.2 was found to enhance receptor binding [28]. However, the mutation reverted in BA.2.75 and its XBB sublineages as well as in BA.2.86 ant its JN.1 sublineages. The reversal reduced receptor-binding affinity but was allowed in the context of the overall strong binding of the BA.2.75 and BA.2.86 lineages for the purpose of immune evasion. Beside Q493, six other amino acids in the RBD are known to experience “Yo-Yo” mutations. Notably, G446 and N501 had mutated between two forms three times, and L452 four times by August of 2023. Meanwhile, deletion of HV69-70 in the N-terminal domain of the Spike had appeared and disappeared three times within the same time frame [30].
1.3. Natural Restrictions on the Improvement of Receptor-Binding Affinity
Although in vitro evolution produced an RBD with a 1000-fold increase in receptor-binding affinity, the highest affinity we have observed in natural variants was no more than 20-fold stronger than wild type. The strongest receptor-binding virus in various reports was BA.2.75, XBB.1.5, or BA.2.86, depending on measurement methods and samples analyzed [14,19,20,21,24,31]. Figure 1F shows dissociation constants (Kd) of the major variants according to and [20], with lower Kd indicating higher binding affinity. The discrepancy between in vitro evolution and the actual in vivo outcomes attests to the differences between isolated molecular interactions on the surface of yeast cells and natural selection of replicating viruses with competing functional priorities in the host cell and the need to compromise under immune pressure. For example, the D614G mutation, which became dominant globally within the first few months of the pandemic [32], resulted in an RBD that bound less tightly to ACE2 than wild type, but the mutation increased the density of intact Spike trimers on the viral surface by preventing premature dissociation of S1 from S2 following furin cleavage in the producing cell [33,34]. As discussed earlier, after high-affinity RBDs were produced, they were quickly deoptimized by immune escape mutations that gave the lineages bigger advantages in human populations. The current trend shows an overall tendency of decrease in receptor-binding affinity (Figure 1F) [20,21,25,35].
Of the nine RBD mutations in the optimal in vitro evolution product, RBD-62 [2], five of them are found in the current LP.8.1* lineage. Interestingly, although V445K in RBD-62 has never been reported in nature, mutation of V445 to another basic amino acids, histidine, has been fixed in the BA.2.86/JN.1 lineage. Recently, H445 was replaced by the third basic amino acid, arginine, in LP.8 and enhanced its receptor-binding affinity [35]. Another mutation in RBD-62, I468F, was found in early variants but was not fixed because it rendered the virus more susceptible to antibody neutralization [36]. The other two mutations in RBD-62 (I358F and T470M) have never been reported in natural variants, suggesting that these residues must be conserved for effective viral replication or for infection of the human host. Two mutations found in another in vitro evolution product with improved ACE2-binding (RBD-71), R408D and K417V, have not been reported in nature, but R408S and K417N/T have been fixed in multiple lineages. The fact that the mutations are tolerated suggests that chances of R408D and K417V emerging in the future exist, but they are unlikely because aspartic acid (D) is categorically different from serine (S), so is valine (V) from asparagine (N) and threonine (T). Nevertheless, it is worthwhile to analyze the impact of D408 and V417 in the context of the current JN.1 sublineages.
2. Deleterious Mutations Lead to Degeneration and Attenuation
Even with enormous population sizes, exponential growth, and strong natural selection, viruses still experience significant genetic drift [37]. Viruses are known to be subject to Muller’s ratchet, the process of irreversible accumulation of deleterious mutations due to random genetic drift, especially in asexual organisms, leading to reduced fitness [37,38]. Beside the high error rates of viral RNA-dependent RNA polymerases (RdRp) [39], mammalian cells use mutagenesis as a defense mechanism to dampen viral fitness [3,4,5,38]. All the Variants of Concern (VOC) derived directly from the ancestral virus instead of evolving from a previous VOC, suggesting lowered adaptability of the variants compared to the ancestral virus. The ancestral virus and most of the early variants are long gone from the human population, leaving the extant subvariants at the mercy of Muller’s ratchet, even with occasional recombination between circulating lineages.
2.1. Declining Mutational Fitness Effects
Bloom and Neher estimated the fitness effects of SARS-CoV-2 mutations by comparing independent occurrences of each mutation to an expected number based on mutation rates at the third nucleotide of four-fold degenerate codons. They found that the overall effects of nonsynonymous mutations in each gene were negative. Moreover, plotting their calculated average fitness effects of nonsynonymous mutations in the RdRp gene and in the S gene over time, we show that the overall fitness effects of missense mutations declined during the reign of Omicron from the beginning of 2022 to the end of 2024 (Figure 1E), which is consistent with the observation of Maiti et al of a dn/ds decline within the Omicron VOC [8]. In other words, further mutations are more and more likely detrimental to viral fitness than being adaptive.
The large genome of SARS-CoV-2 contains regions where natural selection is relaxed. Bloom and Neher’s study also found that fitness effects of nonsynonymous mutations in the accessory proteins were comparable to those of synonymous mutations, i.e., near neutral, consistent with the observed accumulation of missense and nonsense mutations in these genes [8,40].
2.2. Declining Codon Adaptation Index in the Human Host
Beside functional improvements and immune escape, another direction of viral evolution after entering a new host species is optimization of codon usage in the cells of the host, which involves synonymous changes. Selections of nonsynonymous mutations typically take priority over codon usage optimization because the fitness effects of the latter are more subtle. Therefore, it is conceivable to expect sacrifices in codon usage in genes where selective pressure for adaptive evolution is high. Codon adaptation index (CAI) of SARS-CoV-2 in the human population experienced a decline in the early variants, restored in Omicron [41,42,43,44,45], and decreased again [44]. After analyzing the CAI and the codon pair adaptation index (CPAI), Padhiar et al reported no drastic net changes in SARS-CoV-2 genes in codon usage from December of 2019 to July of 2024. However, when we plotted the temporal curves with the supplementary data of Padhiar et al, we found that the S gene experienced two phases of decline in both CAI and CPAI with a sharp increase in the middle, and there was an overall negative correlation between CPAI and time (p = 0.0028, Figure 1G) [46]. This suggests that evolution of the S gene not only has to strike compromises between receptor-binding and immune evasion but also must bear the cost of deoptimized codon usage. Timing of the jump in CAI and CPAI in the S gene coincided with the emergence of Omicron. The large number of adaptive mutations and simultaneous optimization of codon usage in the original Omicron variant suggest a unique evolutionary mechanism which is still a puzzle today [38]. Using the same data set, we also observed significant codon degeneration in the ORF1ab (which includes the RdRp gene) and the N gene, where multiple adaptive mutations have been documented [40,47,48,49]. The CPAI of the whole genome also showed a decreasing trend, although not statistically significant (p = 0.15).
2.3. Phenotypical Attenuation
Accumulation of deleterious synonymous and nonsynonymous mutations resulted in attenuation of SARS-CoV-2 as the pandemic unfolded. There was a decline in replication efficiency in cell cultures from the early B.1.1 variant to Omicron [26,50]. The Omicron subvariants, including some dominant XBB family members, BA.2.86, and JN.1, were less pathogenic in hamsters and in mice than the ancestral B.1 variant [24]. Later Omicron subvariants were less virulent than earlier Omicron subvariants, although BA.2.86 and JN.1 demonstrated more efficient replication in human nasal epithelial cells [22,24].
Attenuation of SARS-CoV-2 in humans manifested as reduced mortality rates over time. There seemed to be a rapid decline in mortality rate during the first few months of the pandemic [51]. COVID-19 related mortality in the United States has been declining continuously even though viral prevalence remains high as evidenced by high test positivity rates (Figure 1H) [52]. The mortality decrease does not correlate with vaccination efforts, as intensive vaccination in 2021 failed to prevent the death peaks later in the year, and the federal government completely stopped free vaccination in September of 2023 [53]. The mortality rate correlates partially with natural immunity. After the Omicron wave in the winter of 2021-2022, most Americans had turned seropositive [54], and mortality dropped dramatically ever since. However, the subsequent gradual decline from 2022 till now probably has more to do with viral attenuation than with herd immunity because of declining vaccine coverage and rapid immune escape of the virus.
A global meta-analysis revealed that the global case fatality rates of the ancestral virus, the Alpha, Beta, Gamma, Delta, and Omicron VOCs were 3.64%, 2.62%, 4.19%, 3.60%, 2.01%, and 0.70%, respectively [55]. Note that the more lethal Beta and Gamma variants emerged before Alpha, and Delta was not as virulent as initially reported in Asia [56]. The case fatality rates of the ancestral virus, the Alpha, Delta, and Omicron VOCs in North America were 4.77%, 2.67%, 2.50%, and 0.73%, respectively, which is roughly consistent with Figure 1H considering the lag time between infection and death and the different prevalence rates of the variants [55]. Viral attenuation and immunity buildup probably both contributed to the decrease in case fatality rate from the original virus to Omicron.
Conclusion
Right now, in the sixth year of the COVID-19 pandemic, there are increasing signs indicating that adaptive evolution of SARS-CoV-2 has plateaued, as reflected in slower accumulation of fixed mutations, recurrent and cyclic mutations at the same sites, declining mutational fitness effects, declining receptor-binding affinity of the Spike protein, deoptimizing codon usage in the human host, and decreasing mortality rates. Moreover, evolution of SARS-CoV-2 has been gradual since the JN.1 sweep early in 2024, and there has only been one addition to WHO’s Variants under Monitoring (VUM, i.e., LP.8.1*) since September of 2024. The last addition to the Variants of Interest (VOI) was JN.1 in December of 2023 and the last addition to the Variants of Concern (VOC) was Omicron (B.1.1.529) in November of 2021. Currently, LP.8.1.1 is posed to slowly displace the other JN.1 sublineages to dominate in the future months. Like its predecessors, the new variant is expected drift under immune pressure as well as Muller’s ratchet.
SARS-CoV-2 may become like the four current human coronaviruses, which cause common cold [57]. Clinically, the symptoms of COVID-19 are increasingly flu-like, and they may become more cold-like in the future.
Acknowledgments
The author thanks Dr. Michael Price for critical reading of the manuscript.
Conflict of Interest
Author declares no conflict of interest.
References
- Carossino M, Izadmehr S, Trujillo JD, Gaudreault NN, Dittmar W, Morozov I, et al. ACE2 and TMPRSS2 distribution in the respiratory tract of different animal species and its correlation with SARS-CoV-2 tissue tropism. Microbiol Spectr 2024;12(2):e0327023. [CrossRef]
- Zahradník J, Marciano S, Shemesh M, Zoler E, Harari D, Chiaravalli J, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol 2021;6(9):1188-1198. [CrossRef]
- De Maio N, Walker CR, Turakhia Y, Lanfear R, Corbett-Detig R, Goldman N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol Evol 2021; 13(5):evab087. [CrossRef]
- Colson P, Chaudet H, Delerce J, Pontarotti P, Levasseur A, Fantini J, et al. Role of SARS-CoV-2 mutations in the evolution of the COVID-19 pandemic. J Infect 2024;88(5):106150. [CrossRef]
- Simmonds P. C→U transition biases in SARS-CoV-2: still rampant 4 years from the start of the COVID-19 pandemic. mBio. 2024;15(12):e0249324. [CrossRef]
- Bloom JD, Neher RA. Fitness effects of mutations to SARS-CoV-2 proteins. Virus Evol 2023;9(2):vead055. Erratum in: Virus Evol. 2024 Mar 26;10(1):veae026. [CrossRef]
- Lippi G, Henry BM. The landscape of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomic mutations. J Lab Precis Med 2022; 7:10. [CrossRef]
- Maiti AK. Progressive evolutionary dynamics of gene-specific ω led to the emergence of novel SARS-CoV-2 strains having super-Infectivity and virulence with vaccine neutralization. Int J Mol Sci 2024; 25(12):6306. [CrossRef]
- Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581(7807):215-220. [CrossRef]
- Gangavarapu K, Latif AA, Mullen JL, Alkuzweny M, Hufbauer E, Tsueng G, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat Methods 2023. [CrossRef]
- Wang X, Hu M, Liu B, Xu H, Jin Y, Wang B, Zhao Y, Wu J, Yue J, Ren H. Evaluating the effect of SARS-CoV-2 spike mutations with a linear doubly robust learner. Front Cell Infect Microbiol. 2023 Apr 19;13:1161445. [CrossRef] [PubMed]
- Dejnirattisai W, Huo J, Zhou D, Zahradník J, Supasa P, Liu C, et al. SARS-CoV-2 Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. Cell 2022;185(3):467-484.e15. [CrossRef]
- Ito J, Suzuki R, Uriu K, Itakura Y, Zahradnik J, Kimura KT, et al. Convergent evolution of SARS-CoV-2 Omicron subvariants leading to the emergence of BQ.1.1 variant. Nat Commun 2023;14(1):2671. [CrossRef]
- Liu H, Wei P, Kappler JW, Marrack P, Zhang G. SARS-CoV-2 Variants of concern and variants of interest receptor binding domain mutations and virus infectivity. Front Immunol 2022;13:825256. [CrossRef]
- Wang Q, Mellis IA, Ho J, Bowen A, Kowalski-Dobson T, Valdez R, et al. Recurrent SARS-CoV-2 spike mutations confer growth advantages to select JN.1 sublineages. Emerg Microbes Infect 2024;13(1):2402880. [CrossRef]
- Jian F, Feng L, Yang S, Yu Y, Wang L, Song W, et al. Convergent evolution of SARS-CoV-2 XBB lineages on receptor-binding domain 455-456 synergistically enhances antibody evasion and ACE2 binding. PLoS Pathog 2023;19(12):e1011868. [CrossRef]
- Taylor AL, Starr TN. Deep mutational scanning of SARS-CoV-2 Omicron BA.2.86 and epistatic emergence of the KP.3 variant. Virus Evol 2024;10(1):veae067. [CrossRef]
- Starr TN, Greaney AJ, Hannon WW, Loes AN, Hauser K, Dillen JR, et al. Shifting mutational constraints in the SARS-CoV-2 receptor-binding domain during viral evolution. Science. 2022 Jul 22;377(6604):420-424. [CrossRef]
- Liu C, Zhou D, Dijokaite-Guraliuc A, Supasa P, Duyvesteyn HME, Ginn HM, et al. A structure-function analysis shows SARS-CoV-2 BA.2.86 balances antibody escape and ACE2 affinity. Cell Rep Med 2024;5(5):101553. [CrossRef]
- Yang H, Guo H, Wang A, Cao L, Fan Q, Jiang J, et al. Structural basis for the evolution and antibody evasion of SARS-CoV-2 BA.2.86 and JN.1 subvariants. Nat Commun 2024;15(1):7715. [CrossRef]
- Yang S, Yu Y, Xu Y, Jian F, Song W, Yisimayi A, et al. Fast evolution of SARS-CoV-2 BA.2.86 to JN.1 under heavy immune pressure. Lancet Infect Dis. 2024 Feb;24(2):e70-e72. Erratum in: Lancet Infect Dis. 2024 Mar;24(3):e156. [CrossRef]
- Tamura T, Mizuma K, Nasser H, Deguchi S, Padilla-Blanco M, et al. Virological characteristics of the SARS-CoV-2 BA.2.86 variant. Cell Host Microbe. 2024;32(2):170-180.e12. [CrossRef]
- CDC: An official website of the United States government [internet]. Atlanta: Center of Disease Control and Prevention; 2024. Update on SARS-CoV-2 Variant BA.2.86 Being Tracked by CDC; 2024 Nov 27 [Cited 2025 Apr 3]. Available from: https://www.cdc.gov/ncird/whats-new/covid-19-variant-update-2023-11-27.html.
- Liu Y, Zhao X, Shi J, Wang Y, Liu H, Hu YF, et al. Lineage-specific pathogenicity, immune evasion, and virological features of SARS-CoV-2 BA.2.86/JN.1 and EG.5.1/HK.3. Nat Commun 2024;15(1):8728. [CrossRef]
- Liu J, Yu Y, Jian F, Yang S, Song W, Wang P, et al. Enhanced immune evasion of SARS-CoV-2 variants KP.3.1.1 and XEC through N-terminal domain mutations. Lancet Infect Dis 2025;25(1):e6-e7. [CrossRef]
- Li P, Faraone JN, Hsu CC, Chamblee M, Liu Y, Zheng Y-M, et al. Neutralization and spike stability of JN.1-derived LB.1, KP.2.3, KP.3, and KP.3.1.1 subvariants. mBio. 2025:e0046425. [CrossRef]
- Chakraborty C, Bhattacharya M. FLip mutations (L455F + F456L) in newly emerging VOI, JN.1: Its antibody and immune escape. Int Immunopharmacol. 2024;133:112146. [CrossRef]
- Philip AM, Ahmed WS, Biswas KH. Reversal of the unique Q493R mutation increases the affinity of Omicron S1-RBD for ACE2. Comput Struct Biotechnol J 2023;21:1966-1977. [CrossRef]
- Mannar D, Saville JW, Poloni C, Zhu X, Bezeruk A, Tidey K, et al. Altered receptor binding, antibody evasion and retention of T cell recognition by the SARS-CoV-2 XBB.1.5 spike protein. Nat Commun 2024;15(1):1854. [CrossRef]
- Focosi D, Spezia PG, Maggi F. Fixation and reversion of mutations in the receptor-binding domain of SARS-CoV-2 spike protein. Diagn Microbiol Infect Dis 2024;108(2):116104. [CrossRef]
- [3[1]] Sugano A, Murakami J, Kataguchi H, Ohta M, Someya Y, Kimura S, et al. In silico binding affinity of the spike protein with ACE2 and the relative evolutionary distance of S gene may be potential factors rapidly obtained for the initial risk of SARS-CoV-2. Microbial Risk Analysis 2023; 25: 100278. [CrossRef]
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020 Aug 20;182(4):812-827.e19. [CrossRef]
- Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 2020;11(1):6013. [CrossRef]
- Zhang J, Cai Y, Xiao T, Lu J, Peng H, Sterling SM, et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021;372(6541):525-530. [CrossRef]
- Liu J, Yu Y, Yang S, Jian F, Song W, Yu L, Shao F, Cao Y. Virological and antigenic characteristics of SARS-CoV-2 variants LF.7.2.1, NP.1, and LP.8.1. Lancet Infect Dis 2025;25(3):e128-e130. [CrossRef]
- [36 Li Q, Wu J, Nie J, Zhang L, Hao H, Liu S, et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020;182(5):1284-1294.e9. [CrossRef]
- Sanjuán R, Domingo-Calap P. Genetic Diversity and Evolution of Viral Populations. Encyclopedia of Virology 2021:53–61.
- Liu Y. Attenuation and Degeneration of SARS-CoV-2 Despite Adaptive Evolution. Cureus 2023;15(1):e33316. [CrossRef]
- Venkataraman S, Prasad BVLS, Selvarajan R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018;10(2):76. [CrossRef]
- Focosi D, Spezia PG, Maggi F. Subsequent Waves of Convergent Evolution in SARS-CoV-2 Genes and Proteins. Vaccines (Basel) 2024;12(8):887. [CrossRef]
- [4[1]] Huang W, Guo Y, Li N, Feng Y, Xiao L. Codon usage analysis of zoonotic coronaviruses reveals lower adaptation to humans by SARS-CoV-2. Infect Genet Evol 2021;89:104736. [CrossRef]
- Posani E, Dilucca M, Forcelloni S, Pavlopoulou A, Georgakilas AG, Giansanti A. Temporal evolution and adaptation of SARS-CoV-2 codon usage. Front Biosci (Landmark Ed) 2022;27(1):13. [CrossRef]
- Mogro EG, Bottero D, Lozano MJ. Analysis of SARS-CoV-2 synonymous codon usage evolution throughout the COVID-19 pandemic. Virology 2022;568:56-71. [CrossRef]
- Fumagalli SE, Padhiar NH, Meyer D, Katneni U, Bar H, DiCuccio M, et al. Analysis of 3.5 million SARS-CoV-2 sequences reveals unique mutational trends with consistent nucleotide and codon frequencies. Virol J 2023;20(1):31. [CrossRef]
- Wu X, Shan KJ, Zan F, Tang X, Qian Z, Lu J. Optimization and Deoptimization of Codons in SARS-CoV-2 and Related Implications for Vaccine Development. Adv Sci (Weinh). 2023;10(23):e2205445. [CrossRef]
- Padhiar NH, Ghazanchyan T, Fumagalli SE, DiCuccio M, Cohen G, Ginzburg A, et al. SARS-CoV-2 CoCoPUTs: analyzing GISAID and NCBI data to obtain codon statistics, mutations, and free energy over a multiyear period. Virus Evol 2025;11(1):veae115. [CrossRef]
- Wang R, Chen J, Gao K, Hozumi Y, Yin C, Wei GW. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun Biol 2021;4(1):228. [CrossRef]
- Wu H, Xing N, Meng K, Fu B, Xue W, Dong P, et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe 2021;29(12):1788-1801.e6. [CrossRef]
- Mears HV, Young GR, Sanderson T, Harvey R, Barrett-Rodger J, Penn R, et al. Emergence of SARS-CoV-2 subgenomic RNAs that enhance viral fitness and immune evasion. PLoS Biol 2025;23(1):e3002982. [CrossRef]
- Mautner L, Hoyos M, Dangel A, Berger C, Ehrhardt A, Baiker A. Replication kinetics and infectivity of SARS-CoV-2 variants of concern in common cell culture models. Virol J 2022;19(1):76. [CrossRef]
- Baud D, Qi X, Nielsen-Saines K, Musso D, Pomar L, Favre G. Real estimates of mortality following COVID-19 infection. Lancet Infect Dis. 2020 Jul;20(7):773. [CrossRef]
- CDC: An official website of the United States government [internet]. Atlanta: Center of Disease Control and Prevention; 2024. Trends in United States COVID-19 Deaths, Emergency Department (ED) Visits, and Test Positivity by Geographic Area; 2024 Apr 7 [Cited 2025 Apr 7]. Available from: https://covid.cdc.gov/covid-data-tracker/#trends_weeklydeaths_testpositivity_00.
- CDC: An official website of the United States government [internet]. Atlanta: Center of Disease Control and Prevention; 2024. COVID data tracker weekly review; 2022 Jul 29 [Cited 2025 Apr 10]. Available from: https://archive.cdc.gov/www_cdc_gov/coronavirus/2019-ncov/covid-data/covidview/past-reports/07292022.html?utm_source=chatgpt.com.
- Clarke KEN, Jones JM, Deng Y, Nycz E, Lee A, Iachan R, et al. Seroprevalence of Infection-Induced SARS-CoV-2 Antibodies - United States, September 2021-February 2022. MMWR Morb Mortal Wkly Re. 2022;71(17):606-608.
- Xia Q, Yang Y, Wang F, Huang Z, Qiu W, Mao A. Case fatality rates of COVID-19 during epidemic periods of variants of concern: A meta-analysis by continents. Int J Infect Dis 2024;141:106950. [CrossRef]
- Ong SWX, Chiew CJ, Ang LW, Mak TM, Cui L, Toh MPHS, et al. Clinical and Virological Features of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.351 (Beta), and B.1.617.2 (Delta). Clin Infect Dis. 2022;75(1):e1128-e1136. [CrossRef]
- Cohen J. Covid’s cold cousins. Science 2024 Jan 12;383(6679):141-145.
Figure 1.
Evolutionary timeline of major SARS-CoV-2 variants. (A) Succession of SARS-CoV-2 variants and Omicron subvariants. Source: GISAID, via CoVariants.org (2025). (B) Phylogenetic relationship of lineages labelled in A. (C) Accumulation of fixed mutations in SARS-CoV-2 over time. Red: all proteins; Blue: the Spike glycoprotein. Mutation counts were obtained from outbreak.info [10]. (D) Average rate of dn/ds change from September 2020 to May 2022. Red: the S gene; Blue: whole genome. Replotted with data from [8]. (E) Temporal change of fitness effects of nonsynonymous mutations from January 2022 to November 2024. Red: the S gene; Blue: the RdRp gene. Plotted with data from Bloom and Neher [6]. (F) Dissociation constants (Kd) of the Spike protein of major SARS-CoV-2 variants. Lower Kd values indicate higher receptor-binding affinity. Data obtained from and [20]. (G) Change in codon pair adaptation index (CPAI) of SARS-CoV-2 between December 2019 and July 2024. Blue: coding sequence of the S gene; Red: all coding sequences. Plotted with data from [46]. (H) Weekly COVID-19 deaths and nucleic acid amplification test percent positivity in the United States from June 2020 to November 2024. Blue: deaths; Orange: test positivity. Data obtained from the Center for Disease Control and Prevention [52].
Figure 1.
Evolutionary timeline of major SARS-CoV-2 variants. (A) Succession of SARS-CoV-2 variants and Omicron subvariants. Source: GISAID, via CoVariants.org (2025). (B) Phylogenetic relationship of lineages labelled in A. (C) Accumulation of fixed mutations in SARS-CoV-2 over time. Red: all proteins; Blue: the Spike glycoprotein. Mutation counts were obtained from outbreak.info [10]. (D) Average rate of dn/ds change from September 2020 to May 2022. Red: the S gene; Blue: whole genome. Replotted with data from [8]. (E) Temporal change of fitness effects of nonsynonymous mutations from January 2022 to November 2024. Red: the S gene; Blue: the RdRp gene. Plotted with data from Bloom and Neher [6]. (F) Dissociation constants (Kd) of the Spike protein of major SARS-CoV-2 variants. Lower Kd values indicate higher receptor-binding affinity. Data obtained from and [20]. (G) Change in codon pair adaptation index (CPAI) of SARS-CoV-2 between December 2019 and July 2024. Blue: coding sequence of the S gene; Red: all coding sequences. Plotted with data from [46]. (H) Weekly COVID-19 deaths and nucleic acid amplification test percent positivity in the United States from June 2020 to November 2024. Blue: deaths; Orange: test positivity. Data obtained from the Center for Disease Control and Prevention [52].
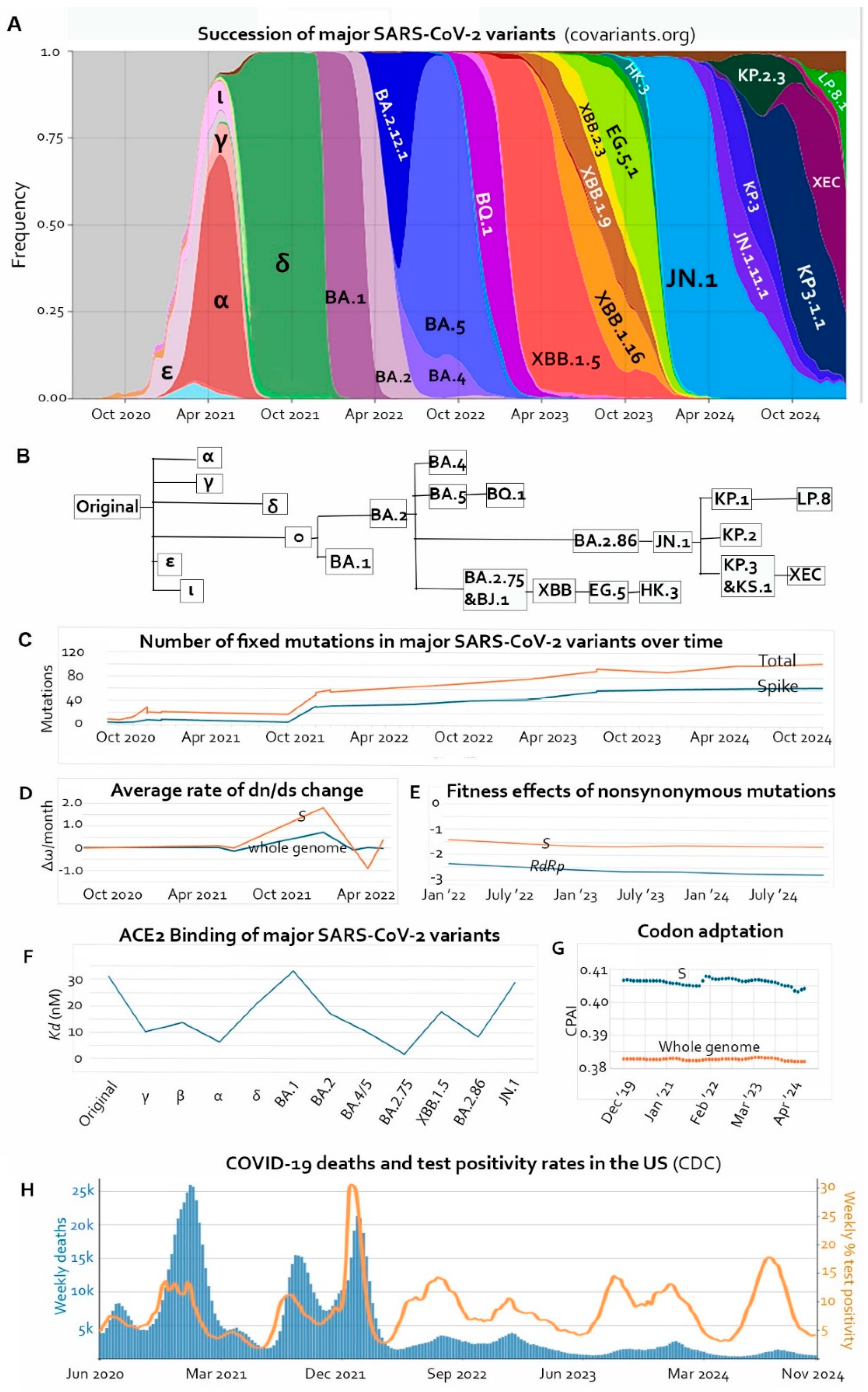

Table 1.
Spike mutations fixed in multiple independent lineages.
| Mutations | Pre-Alpha | Alpha | Beta | Gamma | Delta | Lambda | Omicron |
|---|---|---|---|---|---|---|---|
| HV69-70del | B.1.258, B.1.375 | x | BA.1 | ||||
| Y144del | x | XBB.1.5 | |||||
| R346 | BQ.1.1, XBB.1.5 | ||||||
| K417N/T | x | x | x | ||||
| G446S | BA.1, BA.2.75 | ||||||
| L452R/Q | x | x | BA.4/5 | ||||
| F456L | Multiple XBB and JN.1 sublineages | ||||||
| T478K | x | x | |||||
| E484A/K | x | x | x | ||||
| F490S | x | XBB.1.5 | |||||
| N501Y | x | x | x | x | |||
| D614G | A and B.1 | ||||||
| P681R/H | x | x | x |
Presence in all subvariants is indicated with “x”. If present only in some subvariants, names of selected subvariants are given.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



