
Submitted:
18 April 2025
Posted:
22 April 2025
Read the latest preprint version here
Abstract
We present termal, a fast, interactive terminal-based viewer for multiple se- quence alignments (MSAs), designed for use on remote systems such as high-performance computing (HPC) clusters. Unlike traditional graphical viewers, termal runs entirely within a terminal and offers features such as scrolling, zooming, consensus/conservation visualization, and customizable colour schemes. It is implemented in Rust, ensuring high performance and minimal dependencies.
Keywords:
multiple sequence alignment
; viewer
; terminal
; text user interface
Introduction
Visualising multiple sequence alignments (MSAs) is a common task in computational biology. Many alignment viewers have a graphical user interface (GUI) and are hence unsuitable for use on headless or remote systems such as high-performance computing (HPC) clusters. Command-line tools do exist, for example Alan [1], which stands out as a particularly elegant solution, since it is built on standard Unix tools such as awk and less — indeed, it served as the initial inspiration for the present work. This means, however, that Alan’s interactivity is limited to that of a pager: features such as zooming, reordering sequences, as well as computing and displaying a consensus sequence are absent. While showalign [2] can compute a consensus, it does not support colouring residues, and the user must explicitly call a pager in order to scroll through the alignment. Other programs like alen [3] are interactive, but not all have built-in residue colour schemes or the ability to visually represent metrics such as similarity to the consensus, or to reorder sequences according to such metrics. The capacity to fit a large alignment on screen, typically by only displaying a subset of the sequences and columns, is also rare (see also Table 1). In summary, text-based MSA viewers collectively provide a substantial range of functions, but no viewer implements all, or even most, of them. In this work we introduce termal, which combines most of these features in a single application.
Interface
Apart from the alignment sequences, which occupy the main pane, termal also displays sequence labels and ordinal numbers, a consensus sequence, and a conservation bar plot; it also displays sequence metrics such as similarity to the consensus, or (ungapped) length (Figure 1). The alignment can be scrolled one sequence/column at a time using arrow keys or Vim-like h, j, k, and l; similar keystrokes allow jumping by screenfuls or to the edges of the alignment.
By default, residues of nucleotide alignments are coloured according to Jalview’s [4] nucleotide colour scheme, while protein alignments use that of ClustalX [5]. An alternative colour scheme for protein is Lesk’s [6], and all alignments can be rendered in monochrome.
Alignments too wide to fit on the screen can be "zoomed out" by showing only the first and last column, as well as a sample of equidistant columns in between. The same can be done with sequences for alignments that are too tall. This allows regions of high conservation to be spotted without scrolling. A variant of the zoomed-out mode preserves the alignment’s aspect ratio, at the cost of some wasted space.
The sequences can be reordered according to the currently-displayed sequence metric, in increasing or decreasing order. This allows e.g. to group the most complete sequences together, or those that best match the consensus.
The width of the label pane can be adjusted to fit label length, and both the side and bottom panes can be hidden to maximise the space allocated to the alignment.
termal comes with a built-in help screen that lists all key bindings.
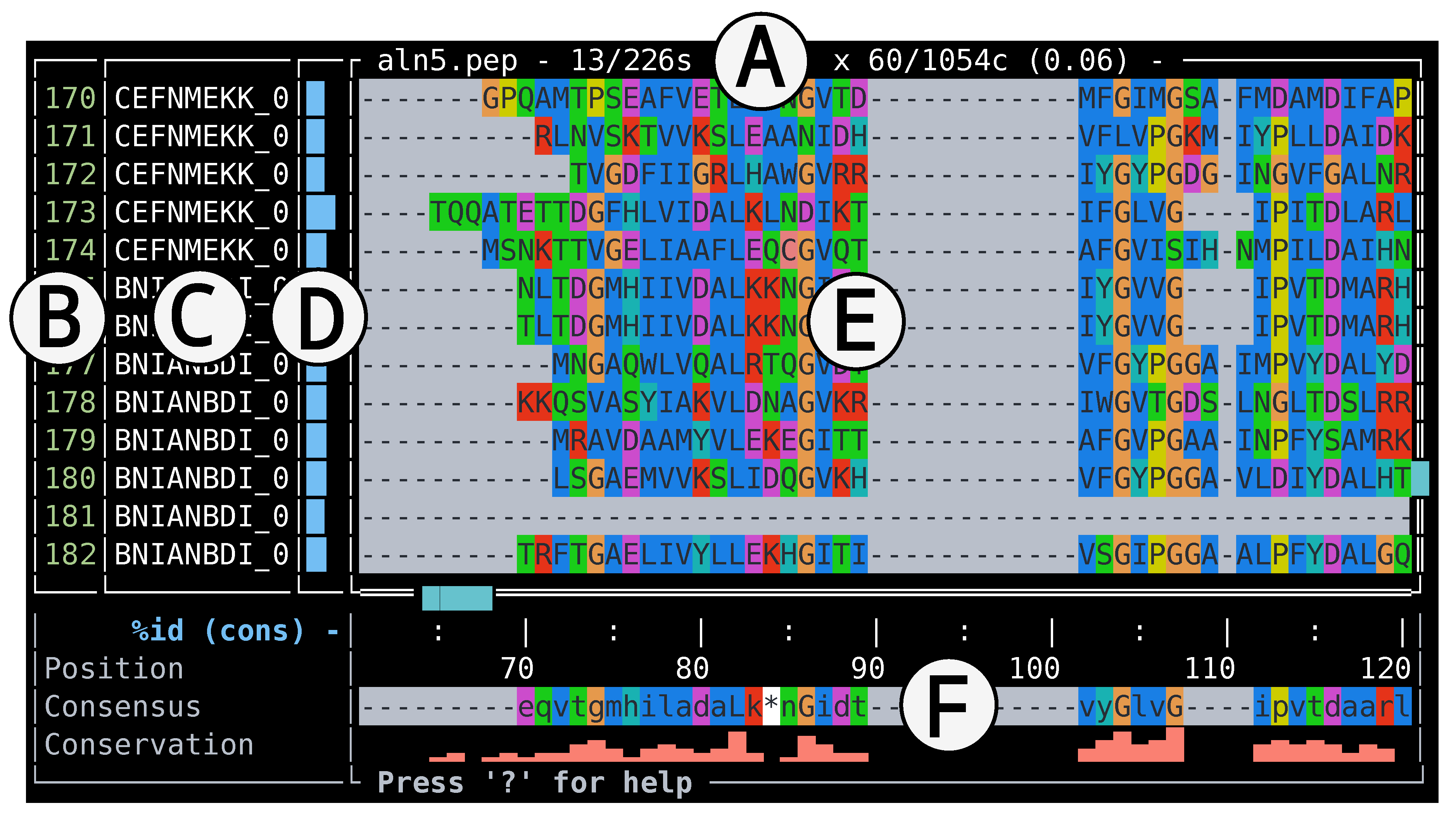
Figure 1.
A snapshot of termal’s interface showing a protein alignment. A: alignment filename and dimensions, B: sequence numbers pane, C: sequence labels pane, D: metric bar plot pane (currently displaying sequence similarity with the consensus), E: alignment pane, F: bottom pane, displaying sequence position, consensus, and conservation bar plot. In this example the sequences are in the original (file) order, and use the CLUSTALX colour scheme. The view is zoomed in, that is, only a fraction of the alignment is displayed.
Figure 1.
A snapshot of termal’s interface showing a protein alignment. A: alignment filename and dimensions, B: sequence numbers pane, C: sequence labels pane, D: metric bar plot pane (currently displaying sequence similarity with the consensus), E: alignment pane, F: bottom pane, displaying sequence position, consensus, and conservation bar plot. In this example the sequences are in the original (file) order, and use the CLUSTALX colour scheme. The view is zoomed in, that is, only a fraction of the alignment is displayed.
Performance and Limitations
termal has been tested on alignments exceeding 15,000 sequences and 1,500 columns ( 22 million alignment cells), with startup and initial rendering completing in under one second on a machine with 12th-generation Intel® CoreTM i5-1240P CPU and 16 GB of RAM running Linux 6.14.2. In practice, interactive performance is limited more by the speed of the terminal emulator than by termal itself. GPU-accelerated terminals such as Alacritty [7], Kitty [8] and Ghostty [9] offer smoother scrolling at large screen sizes than do more traditional emulators.
Comparison with Existing Tools
Table 1 presents a feature matrix for termal and a selection of other TUI alignment viewers.
Availability
termal is distributed under the MIT license. It is available as a single precompiled binary (for Linux, MacOS, and Windows), with no external dependencies or runtime environment required, from gitlab.sib.swiss/tjunier/termal.git. Alternatively, users with Rust installed can install it via cargo install termal.
Conclusion
While this work is not intended as a comprehensive review of alignment viewers, we surveyed several tools with comparable goals — namely, terminal-native operation and varying degrees of interactivity — including showalign, alan, alv, and alen. To our knowledge, termal is the only tool to combine interactive navigation, zooming, consensus and conservation visualization, several built-in colour maps, as well as display of and ordering by sequence metrics in a single terminal interface. Its minimal dependencies and fast startup make termal suitable for both ad-hoc use and for integration into semi-automated workflows requiring terminal-based alignment review. Accordingly, termal fills a niche for fast, interactive MSA exploration directly in the terminal, making it an ideal tool for remote bioinformatics workflows.
Acknowledgments
The development of termal was funded by Swiss National Science Foundation BRIDGE Discovery grant 40B2-0_194701. The author wishes to thank Drs Guillaume Cailleau and Sébastien Moretti for insightful comments on the program.
References
- Dunne, M. Alan: A lightweight alignment viewer for the terminal. https://github.com/mpdunne/alan, 2020. Accessed 2025-04-13.
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in Genetics 2000, 16, 276–277.
- Nissen, J. alen: Interactive terminal-based MSA viewer. https://github.com/jakobnissen/alen, 2023–2024. Accessed 2025-04-13.
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191.
- Larkin, M.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.; McWilliam, H.; Valentin, F.; Wallace, I.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948.
- Lesk, A.M. Introduction to bioinformatics; Oxford university press, 2019.
- Wilm, J.; contributors. Alacritty - A cross-platform, GPU-accelerated terminal emulator. https://github.com/alacritty/alacritty, 2017–2024. Accessed 2025-04-13.
- Goyal, K. Kitty - The fast, feature-rich, GPU-based terminal emulator. https://sw.kovidgoyal.net/kitty/, 2017–2024. Accessed 2025-04-13.
- Hashimoto, M. Ghostty - A modern, GPU-accelerated terminal emulator. https://github.com/mitchellh/ghostty, 2023–2024. Accessed 2025-04-13.

Table 1.
Feature comparison of terminal-based MSA viewers.
| Feature | termal | alen | alv | alan | showalign |
|---|---|---|---|---|---|
| Basics | |||||
| Language | Rust | Rust | Python | Shell | C |
| Interactivity | Yes (full TUI) | Yes (TUI) | No (pager output) | No (pager output) | No (static output) |
| Handles large alignments | Yes (tested >15k×1500) | Yes | Yes (slower) | Yes (but static) | Moderate |
| Display features | |||||
| Scrolling / navigation | Yes | Yes | No | No | No |
| Zooming | Yes | No | No | No | No |
| Label pane toggle | Yes | No | No | No | No |
| Consensus display | Yes | Yes (reference toggle) | Yes (static) | No | Optional |
| Conservation display | Yes | No | No | No | No |
| Similarity histogram | Yes (vertical bars) | No | No | No | No |
| Colour schemes | Multiple, configurable | Fixed | Fixed | Basic ANSI | Limited |
| Sequence numbering | Yes | No | No | No | Yes |
| Supported formats | FASTA | FASTA | FASTA, Clustal (via BioPython) | FASTA | FASTA, Clustal |
| Sorting / filtering | |||||
| Sort by consensus similarity | Yes | No | No | No | No |
| Sort by sequence length | Yes | No | No | No | No |
| Manual row reordering | No | Yes | No | No | No |
| Regex search | No | Yes | No | No | No |
| Integration / output | |||||
| Output style | TUI | TUI | STDOUT (to pager) | STDOUT (to pager) | Plain text output |
| HPC / CLI friendly | Yes (single binary) | Yes | Needs Python env | Yes | Yes (installed via EMBOSS) |
| License | MIT | MIT | MIT | Various | GPL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



