
Submitted:
21 April 2025
Posted:
27 April 2025
You are already at the latest version
Abstract
Metastatic cancer is the final frontier in disease progression with few avenues of treatment open to patients. The failure of therapeutic options at the end-stage disease is confounded by the presence of diverse clones in the metastatic tumor which contributes to intratumoral heterogeneity (ITH) resulting in intrinsic and acquired drug resistance. We aim to elucidate the contribution of ITH to the development of metastasis with a focus on the origins and molecular mechanisms driving ITH, as well as the clinical and technical challenges of acquiring and studying metastatic cohorts in which we can investigate this phenomenon. Bioinformatic approaches that could help in silico analysis will be discussed which could shed light on how to design new therapeutic strategies for metastatic cancer and give hope to patients living under the shadow of the sword of Damocles.
Keywords:
intratumoral heterogeneity
; metastasis
; drug resistance
; tumor microenvironment
; single-cell profiling
; spatial profiling
1. Introduction
Cancer is a formidable global health concern, claiming 9.7 million lives in 2022 [1]. Despite the significant research efforts and financial investments in managing cancer patients, instances of cancer relapses and treatment failure continue to persist. One of the major causes for poor prognosis and increased mortality rates among cancer patients is metastasis [2]. A SEER database study revealed that from 1992 to 2019, 1 million patients were diagnosed with metastatic cancer and 80.9% succumbed to the disease. This underscores the immense burden of metastasis, a complex multistep process that involves migration of cells from the primary tumor to local or distant parts of the body, giving rise to secondary tumors [3,4]. To survive the fraught metastatic journey, tumor cells undergo a series of changes which includes loss of cell-cell adhesion, acquisition of mesenchymal characteristics, entry into microvasculature of the circulatory systems and successful colonization at a secondary site. Concurrently, metastatic cells also interact with the components of tumor microenvironment (TME) and evade immune surveillance of the host immune system. Due to these immense selective pressures, only a small subset of cells within the primary tumor successfully metastasize to distant sites [4]. Alongside tumor evolution, the metastatic potential of selective cancer cells is determined by intratumoral heterogeneity (ITH) [5,6].
ITH refers to the presence of distinct cancer cell populations within a tumor that exhibit different phenotypic and genotypic properties as a result of intrinsic and extrinsic factors [7]. Intrinsic factors include genetic mutations, transcriptomic, epigenetic and translational modifications that help in remodeling the cellular machinery of tumor cells, giving rise to distinct phenotypes. On the other hand, extrinsic factors are the components of the TME like stromal cells, cancer-associated fibroblasts (CAF), tumor-associated macrophages (TAM) and signaling molecules (chemokines, cytokines), which influence immune cell activity, create hypoxic conditions, alter vasculature, affect tumor cell metabolism and help establish a pre-metastatic niche [7]. Together, these factors significantly contribute to mechanisms driving metastasis [8,9]. An example of a prominent biological program is epithelial-mesenchymal transition (EMT), that enables selected epithelial cancer cells to acquire mesenchymal properties, like enhanced migratory capacity and invasiveness that are critical for the metastatic process. Among the heterogenous tumor populations, the ability of a few epithelial cells to undergo EMT is dependent on the expression of transcription factors (SNAIL, TWIST, and ZEB), proteins (N-Cadherin, Vimentin), hypoxia inducible factors (HIFs) and cytokines (like TGF-β) [8]. Therefore, the selection of these rare, metastatic cells, is strongly influenced by the inherent property of ITH. Furthermore, ITH is implicated in giving rise to distinct phenotypes within the metastatic cells. In a particular study, Brown et al. isolated single-cell clones from the SUM149PT human breast cancer cell line and evaluated the metastatic ability of cancer cells, using a multiomics approach. Findings of the study revealed that the cancer cells exhibited an EMT spectrum, which included subpopulations depicting epithelial (E), intermediate EMT (EM1, EM2, EM3) and mesenchymal (M1, M2) phenotype. The intermediate EMT cells characterized by the increased expression of CBFβ protein, exhibited a 2- to10-fold higher migratory and invasive ability than the mesenchymal cells. Furthermore, lung metastases obtained from surgical resection of mice injected with the SUM149PT cell line, revealed different EMT phenotypes contributed by intermediate EMT cells as opposed to epithelial and mesenchymal clones. The EM1 formed a mixture of micro and macrometastases, whereas the EM2 and EM3 phenotypes predominantly contributed to micrometastases and macrometastases respectively. This study clearly demonstrates that phenotypic heterogeneity within EMT states can impact the metastatic progression and disease outcomes [9].
ITH is also recognized as a leading cause of poor prognosis and therapeutic failure among metastatic patients [10]. The heterogeneity within a tumor creates subpopulations exhibiting varying drug sensitivities, that escape the effects of cancer therapy, resulting in the survival of rare, resistant cells [10,11,12,13]. An example of ITH contributing to drug resistance is illustrated by a study [12], which evaluated the dynamics of drug-resistant cells among colorectal cancer (CRC) patients. Single-cell RNA sequencing (scRNA-seq) and Fluorescence activated cell sorting (FACS) of 2-dimensional patient organoids (2Do) revealed heterogeneous populations of cells (POU5F1-positive and POU5F1-negative cells) exhibiting differential expression of POU5F1. In vitro administration of anticancer drugs to 2DOs showed a higher proportion of chemo-resistant POU5F1-positive cells towards the end of drug treatment as compared to the POU5F1-negative cells. While POU5F1-negative cells showed no liver metastases (0/4), POU5F1-positive cells demonstrated a significantly higher metastatic potential (4/4), along with upregulation of the Wnt/β-catenin signaling pathway. Treatment with Wnt/β-catenin pathway inhibitor called XAV939, notably reduced β-catenin expression in the chemoresistant POU5F1-positive cells and lead to tumor shrinkage, suggesting the pivotal role of ITH in the development of drug resistance and underscores the potential of targeted therapy among metastatic patients.
While considerable research has highlighted the role of intratumoral heterogeneity (ITH) in metastasis and drug resistance, there remains significant gaps in understanding the molecular mechanisms of ITH and its prominent contribution to metastatic progression and treatment failure. This review aims at improving our understanding of metastatic ITH, addressing the technical challenges in the field and exploring new avenues of treatment for managing metastasis.
2.1. Intratumoral Heterogeneity Promotes the Metastatic Cascade
ITH as a phenomenon, has been corroborated across cell lines, animal models and clinical cohorts in various metastatic cancers including breast cancer, lung cancer, colorectal cancer, liver cancer, prostate cancer and melanoma [9,12,14,15,16]. For instance, Fujino et al., identified two distinct subpopulations with varying metastatic potentials (POU5F1-low and POU5F1-high cells) in 2D organoids obtained from colorectal cancer patients [12]. Peng et al., observed two ductal cell types (type 1 and 2 ductal cells) exhibiting different transcriptomic profiles, in tumor samples obtained from Pancreatic ductal adenocarcinoma (PDAC) patients [15]. These cells differed in gene expression, proliferative rate and migratory potentials. The type 1 ductal cells were relatively normal and present in both non-cancerous and neoplastic tissues, whereas type 2 ductal cells were malignant [13]. Hapach et al., isolated two distinct subpopulations of circulating tumor cells (weakly migratory and highly migratory) in the human metastatic breast cancer cell lines MDA-MB-231 based on migration abilities [16]. A computational study on CRC liver metastases revealed five distinct clusters of de novo liver metastasis subtypes (LMS1 – LSM5) which exhibited different EMT phenotypes, based on gene expression (like TP53, KRAS, NRAS, and BRAF) [9]. These studies highlight the contribution of ITH in metastatic progression and emphasize the need to study factors mediating ITH in metastatic cells.
ITH can be studied at genetic, transcriptomic, epigenetic and post-translational levels, all of which contributes to the underlying intrinsic factors. At the genetic levels, two types of mutations (driver mutations and passenger mutations) can lead to heterogeneity. Driver mutations are genetic alterations that provide a selective advantage to cancer cells and directly contribute to tumor initiation and progression. These mutations are positively selected and emerge as dominant clones in most primary cancers. Examples of driver mutations include variations in genes like TP53, PTEN, PIK3CA, MAPK. On the other hand, passenger mutations do not confer selective advantages but are considered as the main source of clonal diversity in established tumors [13,14]. Together, these mutations contribute to genetic heterogeneity, which is said to play a pivotal role in driving metastasis. A study conducted by Wang et al. analyzed the genomic instability in primary lung adenocarcinoma (LP) and matched brain metastasis (BM). Analysis of Somatic copy number alteration (SCNA) burden revealed a significantly higher genomic instability and upregulation of EGFR and SMAD4 genes in BM (p=3.8e-05) as compared to LP tumors. This indicates that continual accumulation of these genomic instability variants in LP resulted in selection of few metastatic cells seeding BM. Furthermore, genetic ITH score was found to be greater than 1 in 77% patients with BM as compared to LP, which implies genetic heterogeneity expands in the course of metastatic progression [11]. Therefore, analyzing genetic ITH is essential for identifying rare gene variants within a tumor which may contribute to metastasis. Using a multiomics approach, Roper et al. analyzed samples from rapid autopsies of metastatic thoracic tumors (lung adenocarcinoma and thymic carcinoma) and observed an aggressive phenotype characterized by a high percentage of APOBEC3 germline variants. These variants were in turn upregulated by TP53 gene mutations and signals from Interferon gamma (IFN-γ) induced APOBEC expression, which also highlights the role of tumor immune microenvironment in driving ITH [17].
Besides genetic mutations, variations in gene expression profiles can also affect the metastatic potential of tumor cells. By sequencing whole genomes and paired transcriptomes procured from surgical cohorts for hepatocellular carcinoma (HCC), Zhai et al. revealed high level of phenotypic ITH in stage II cancer patients on account of multiple transcriptomic variations. Among the 67 patients, 30% exhibited more than one RNA subtype (mixed transcriptomic subtypes) which showed a more aggressive phenotype (upregulated cell cycle) as compared to the rest HCC patients (downregulated cell cycle and less aggressive phenotype) [18]. Similarly, scRNA-seq of HER2 breast cancer revealed a transcription factor (ZFP281) responsible for two prominent phenotypes within disseminated cancer cells (DCC). Upregulation of ZFP281 in DCCs induced a mesenchymal-like dormant phenotype leading to early lung metastasis outgrowth. Downregulation of ZFP281 and CDH11 resulted in loss of an invasive mesenchymal phenotype within the DCCs [19]. Winkler et al. analyzed the single-cell transcriptomes of primary tumor and matched metastasis from patient-derived xenograft (PDX) models of breast cancer which showed profound transcriptional differences between the tumors. Based on the expression of EMT markers and genes involved in immune regulation, the primary tumor cells were further classified as poorly metastatic (downregulated expression of EMT markers, high immune cell regulation), intermediate metastatic and highly metastatic (upregulated markers of EMT, low immune cell regulation). Furthermore, Kaplan-Meier plotter revealed that the intermediate EMT phenotype, characterized by upregulation of 5 marker genes (CD24, CRYAB, KRT15, CALML5, and S100A2) was particularly correlated with worse patient outcomes (p=1.2e-5) [20]. Therefore, identifying the transcriptomic signatures of this phenotype could help in identifying potential new therapeutic targets to prevent metastatic development.
In recent years, researchers have identified the critical role of epigenetic modifications like altered DNA methylation, histone modifications and chromatin remodeling in influencing gene expression and resulting in distinct phenotypes [13]. For instance, Hua et al., investigated the heterogeneity in SCNAs and DNA methylation among 84 lung adenocarcinoma patients. The degree of ITH within the SCNAs was quantified using a statistical association index (APITH) and the survival risk was estimated using the Cox proportional-hazards model. The APITH index was higher in patients showing high levels of SCNAs and methylation profiles indicating a high level of ITH. Additionally, the Cos proportional-hazard model indicated that patients showing higher methylation profiles characterized by significantly altered CpG island methylator phenotype were associated with risk of distant metastasis (HR = 1.35, 95% CI = 1.07–1.72, p = 0.012) and poor overall survival (HR = 1.27, 95% CI = 1.05–1.55, p = 0.016), highlighting the key role of epigenetic modification in driving metastatic potential [21].
Protein expression is the ultimate goal of gene translation and is said to be a critical source of ITH. Given that proteins are the final effectors of all cellular pathways, they often serve as critical targets for therapy. Therefore, it is essential to understand the heterogeneity in protein expression and its role in metastasis. Based on the expression of protein markers, weakly migratory and highly migratory circulating tumor cells were obtained in the human metastatic breast cancer cell lines MDA-MB-231. Transwell migration assay confirmed that these populations exhibited different metastatic potential. The weakly migratory subpopulation showed higher metastatic ability and were characterized by depletion of a protein called E-cadherin, whereas the highly migratory subpopulation showed lower metastatic ability and was marked by higher E-cadherin expression [16]. This highlights the role of differential protein expression in influencing the migratory and metastatic abilities of tumor cells. In a comprehensive study, high level of heterogeneity among proteins involved in cell-cycle progression was observed in the bone metastases (BM) as compared to prostate cancer. The bone metastases were further classified into two distinct phenotypic subgroups based on differential protein expression. The BM 1 group exhibited higher levels of Prostate-specific antigen (PSA) and low levels of minichromosome maintenance protein 3 (MCM3) whereas the BM 2 group tumors were enriched in cell proliferation processes like DNA replication, mitosis, and mRNA processing and showed high MCM3 and low PSA expression. Kaplan–Meier analysis implied that patients with BM2-like metastases had shorter survival times after first therapy compared to the BM1-like group, indicating that protein heterogeneity in metastatic tumors is critical for therapeutic interventions for prostate cancer [22].
In addition to the intrinsic factors, the role of TME is critical in driving ITH and disease progression. A study conducted by Zou et al., investigated the effect of anti-PD-1/L1 immune checkpoint therapy in metastatic breast cancer sites. Post therapy, a higher percentage of immune resistant cells were found in liver and brain metastasis, which were attributed to the immune TME. The immune metastatic environment reprogrammed the immunosuppressive cells such as FOXP3+ regulatory T cells, LAMP3+ tolerogenic dendritic cells in liver and brain metastases compared to the other sites, leading to chemoresistance [23]. In vivo isotope tracing analysis in PDX of melanoma patients revealed populations exhibiting different levels of lactate and Monocarboxylate transporter 1 (MCT1), impacting the metastatic potential. The efficient metastatic cells showed higher levels of MCT1 and uptake of lactate, whereas inefficient metastatic cells showed inhibition of MCT1 and reduced lactate uptake. Knockdown of MCT1 in mice models resulted in reduced metastatic burden, indicating that MCT1 could also be a potential target for cancer therapy [24]. Similarly, a study on colorectal cancer liver metastases revealed five distinct clusters of de novo liver metastasis subtypes (LMS1 – LSM5) which exhibited different EMT phenotypes, based on gene expression (TP53, KRAS, NRAS, and BRAF). Contrary to findings of other studies, which associate high level of heterogeneity with poor prognosis [9,10,16], Principle Component Analysis confirmed that LMS1, which was the least heterogeneous subtype, exhibited epithelial phenotype and was associated with worse survival outcomes of LMS patients [14]. This could imply that the impact of ITH on metastatic progression is dependent on the cancer-type or external signals from the TME can also impact the disease. While LMS2, LMS3 and LMS4 subpopulations exhibited a transit amplifying phenotype, LMS5 was the only mesenchymal-like subtype, that showed high levels of transcriptomic heterogeneity which was in turn influenced by stromal and immune cell infiltration from the TME [25]. Therefore, in addition to genetic mutations, the interactions between tumor cells and the surrounding TME is equally important in driving phenotypic heterogeneity and outcomes in metastatic patients.
Figure 1.
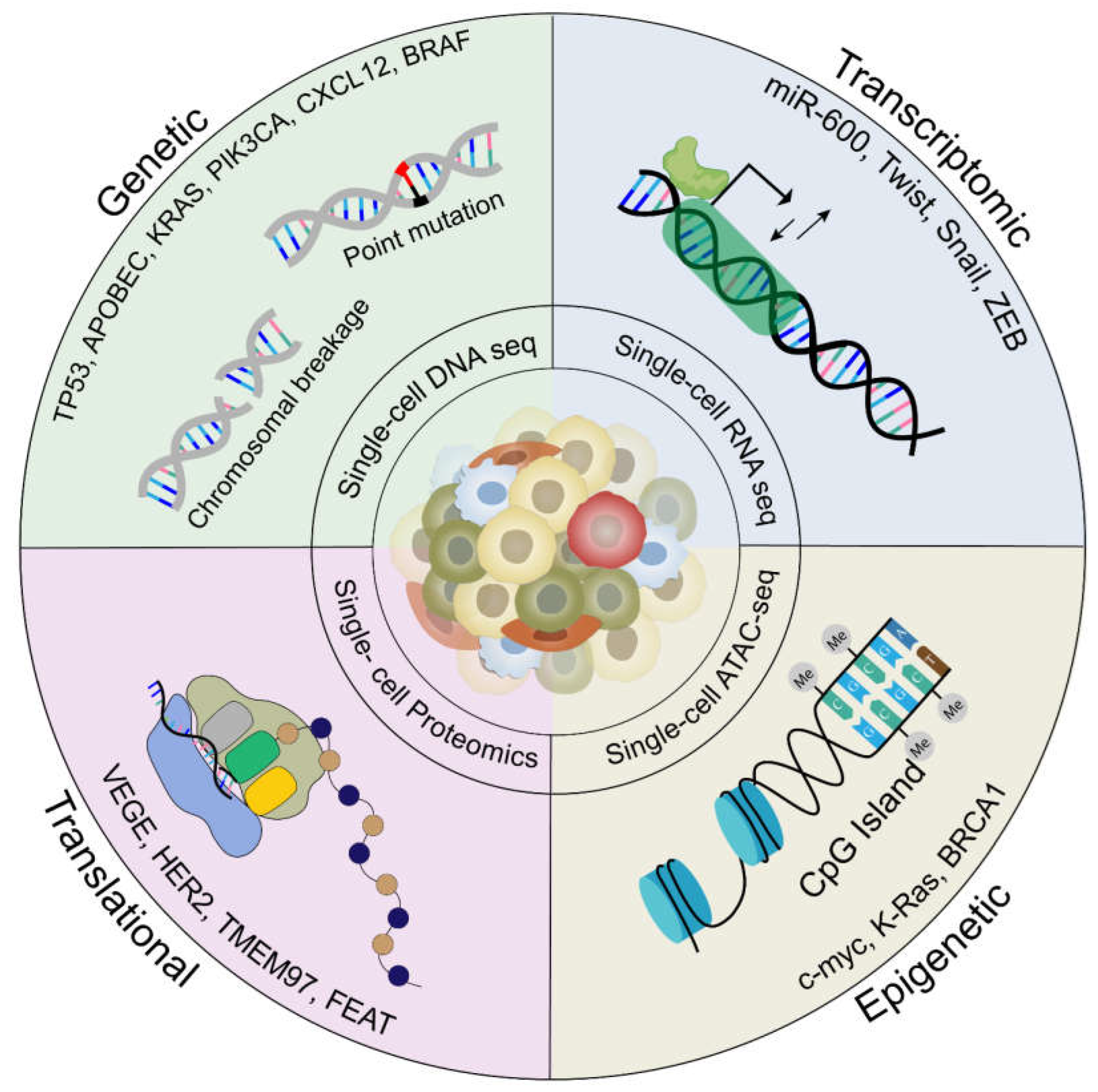
Molecular drivers of intratumoral heterogeneity in metastasis. Intratumoral heterogeneity is governed by four major molecular drivers: Genetic mutations, transcriptomic, epigenetic and translational modifications, which gives rise to distinct phenotypes within tumor cells during metastasis. Genetic mutations include point mutation in genes like TP53, APOBEC , PIK3CA and chromosome breakage in genes like CXCL2 leading to upregulation of prominent metastatic pathways which can be studied using Single-cell DNA sequencing. Transcriptomic modifications include dysregulation of transcription factors like miR-600, TWIST, SNAIL and ZEB, can be studied using Single-cell RNA sequencing. Epigenetic modifications include hypermethylation in the CpG islands of genes like c-myc, KRAS and BRCA1 studied using Single-cell ATAC sequencing and translational modifications dysregulation of proteins VEGF, HER2, TMEM97 and FEAT can be studied using Single-cell Proteomics profiling.
Figure 1.
Molecular drivers of intratumoral heterogeneity in metastasis. Intratumoral heterogeneity is governed by four major molecular drivers: Genetic mutations, transcriptomic, epigenetic and translational modifications, which gives rise to distinct phenotypes within tumor cells during metastasis. Genetic mutations include point mutation in genes like TP53, APOBEC , PIK3CA and chromosome breakage in genes like CXCL2 leading to upregulation of prominent metastatic pathways which can be studied using Single-cell DNA sequencing. Transcriptomic modifications include dysregulation of transcription factors like miR-600, TWIST, SNAIL and ZEB, can be studied using Single-cell RNA sequencing. Epigenetic modifications include hypermethylation in the CpG islands of genes like c-myc, KRAS and BRCA1 studied using Single-cell ATAC sequencing and translational modifications dysregulation of proteins VEGF, HER2, TMEM97 and FEAT can be studied using Single-cell Proteomics profiling.
2.2. Implications of Intratumoral Heterogeneity in Therapeutic Resistance
Drug resistance is a complex, multifaceted phenomenon by which tumor cells develop tolerance to cancer therapy [26]. It is a major factor responsible for cancer relapse and one of the leading causes of cancer related death. Drug resistance is categorized into two types: - intrinsic resistance and acquired resistance. Intrinsic resistance is the inability of the cancer cells to respond to treatment, due to mutations of driver genes as well as selective pressures from the TME [24,26,27,28]. On the other hand, acquired resistance happens when tumor cells develop resistance to the treatment, upon prolonged exposure to the drug, eventually leading to relapse. The tumor cells enter the quiescent phase to escape chemotherapy and develop resistance over time [26]. The major mechanisms leading to the two types of drug resistance include activation of drug efflux pumps, alteration in structures of transporters present in the plasma membrane, interactions from the tumor microenvironment like hypoxic conditions induced by reactive oxygen species (ROS), altered glucose metabolism and increased paracrine signaling of Tumor Associated Macrophages. Few studies have demonstrated how these mechanisms, coupled with the inherent ITH, leads to drug resistance in metastatic cancer [14,16,17,18,19,20,21,25,26,29]. For instance, Ryl et al., suggested that oncogene MYC expression levels influence the response of patient-derived neuroblastoma cells to the first-line chemotherapy, doxorubicin. Post-treatment, two heterogeneous sets of populations (low and high MYC-expressing cells) exhibited different mechanisms of resistance, based on MYC gene levels. The low-MYC cells entered therapy-induced senescence whereas the high-MYC cells continued to proliferate during chemotherapy. The response of high-MYC cells was attributed to position of cells in the cell cycle (G1 phase), before the treatment. This study indicates the role of therapeutic pressures in influencing ITH, leading to the emergence of drug-resistant populations [28]. Similarly, Yang et al. used Shannon index (mathematical model used to measure heterogeneity) to predict the association between ITH and clinical outcomes among Triple-negative breast cancer (TNBC) patients. The ITH of each tumor was characterized by calculating the Copy number variations (CNVs) of Myc, Epidermal growth factor and cyclin D1. Patients suffering from metastatic TNBC showed higher shannon weiner indices for all the biomarkers and was significantly associated with worse metastasis-free survival (MFS) (p<0.05) [30]. McMahon et al. studied the ITH in acute myeloid leukaemia (AML) and found subclones that developed resistance after treatment with FLT3 inhibitors. These clones were characterized by the accumulation of RAS/MAPK mutations [31]. Brady et al. identified a subset of amphicrine tumor cells (AR+/NE+, androgen receptor and neuroendocrine) that were resistant to the classic androgen receptor (AR)-directed therapy for treating metastatic prostate cancer. These cells were characterized by the overexpression of splice variants of the AR, (in particular, AR-V7) as determined by RNA sequencing [32]. Thankamony et al., isolated two tumor cell populations (T1 and T2) exhibiting distinct morphology and phenotypes from primary tumors of 4T1 TNBC mice model. Phenotypic characterization revealed that the T1 cells exhibited higher proliferative ability, reduced self-renewal capacity and were associated with an aggressive disease outcome, compared to the T2 cells [33]. Bioinformatic analyses showed that Metastasis associated colon cancer 1 (Macc1) was one of the top candidate genes mediating the aggressive phenotype in the T1 tumor cells, which could be potentially targeted in the future for improving TNBC treatment regimens [23]. These studies suggest that investigating ITH associated with metastatic phenotypes can help in identifying critical targets for managing metastatic cancer patients.
Figure 2.
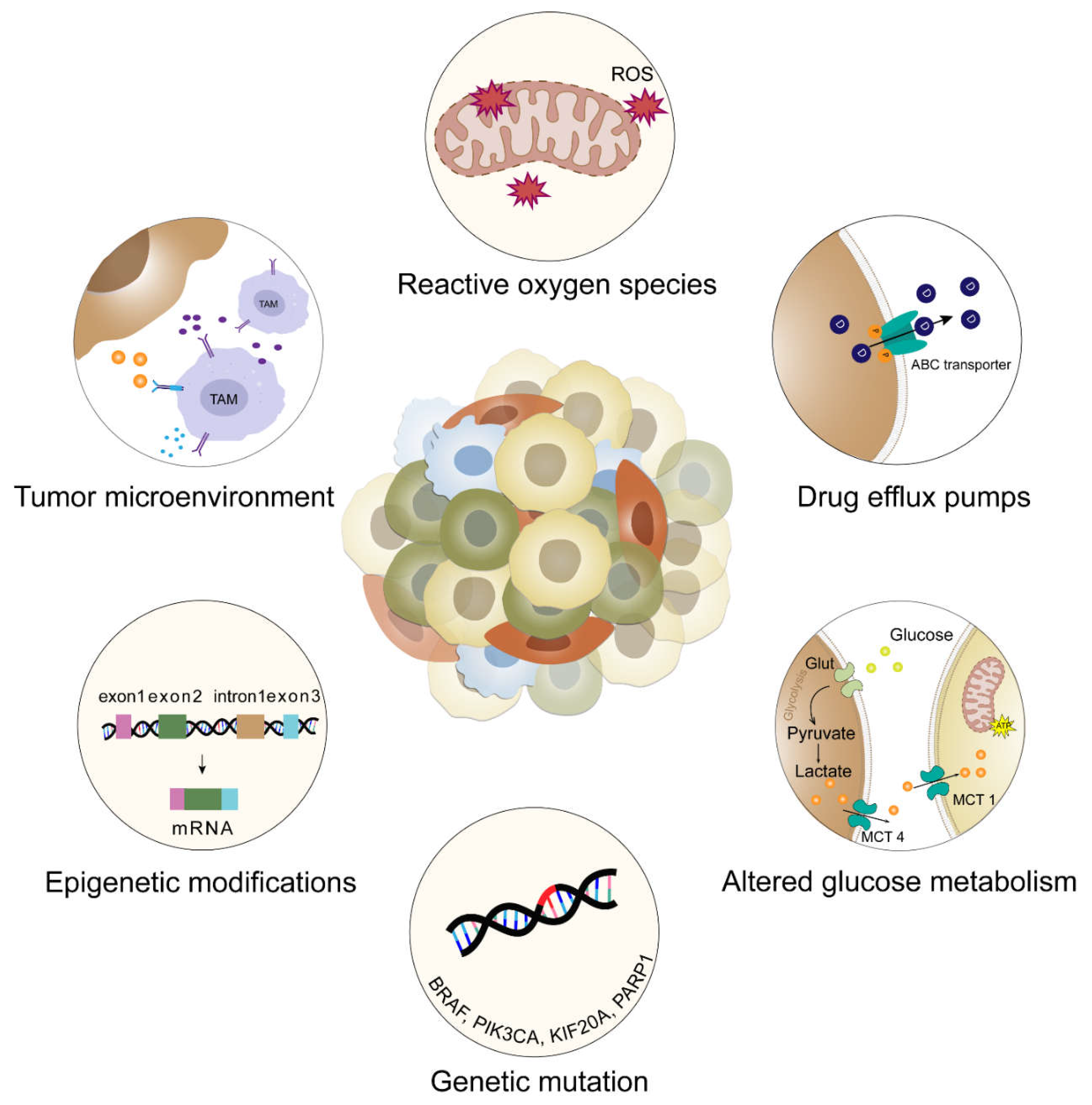
Mechanisms of intrinsic and acquired drug resistance. Drug resistance is classified into two types: clinical intrinsic resistance and clinical extrinsic resistance. Clinical intrinsic resistance is the inability of the cancer cells to respond to treatment, due to mutations of driver genes as well as selective pressures from tumor microenvironment. Clinical acquired resistance occurs when tumor cells develop resistance to the treatment, upon prolonged exposure to the drug, eventually leading to relapse. Several mechanisms leading to the two types of drug resistance are as follows: genetic mutations (BRACA, PIK3CA, KIF20A, PARP1) and epigenetic modifications (e.g. alternate splicing leading to overexpression of splice variants of genes like MCL-1, VEGF) leads to uncontrolled cell division, tumor proliferation, inability of cells to undergo apoptosis and reduced sensitivity to chemotherapy and endocrine therapy. Activation of drug efflux pumps (e.g. ATP-binding cassette [ABC] transporters) and alteration in structures of transporters present in the plasma membrane leads to decreased drug uptake by cancer cells. The ABC transporters actively pump out xenobiotics through ATP hydrolysis and reduce intracellular concentrations, leading to multidrug resistance (MDR) in cancer and treatment failure. Interactions from the TME like hypoxic conditions induced by Reactive oxygen species (ROS), altered glucose metabolism (e.g. altered expression of enzyme like GLUT and upregulation of glycolytic pathways) and increased paracrine signaling of TAMs leads to an immunosuppressive environment, rendering immunotherapy ineffective.
Figure 2.
Mechanisms of intrinsic and acquired drug resistance. Drug resistance is classified into two types: clinical intrinsic resistance and clinical extrinsic resistance. Clinical intrinsic resistance is the inability of the cancer cells to respond to treatment, due to mutations of driver genes as well as selective pressures from tumor microenvironment. Clinical acquired resistance occurs when tumor cells develop resistance to the treatment, upon prolonged exposure to the drug, eventually leading to relapse. Several mechanisms leading to the two types of drug resistance are as follows: genetic mutations (BRACA, PIK3CA, KIF20A, PARP1) and epigenetic modifications (e.g. alternate splicing leading to overexpression of splice variants of genes like MCL-1, VEGF) leads to uncontrolled cell division, tumor proliferation, inability of cells to undergo apoptosis and reduced sensitivity to chemotherapy and endocrine therapy. Activation of drug efflux pumps (e.g. ATP-binding cassette [ABC] transporters) and alteration in structures of transporters present in the plasma membrane leads to decreased drug uptake by cancer cells. The ABC transporters actively pump out xenobiotics through ATP hydrolysis and reduce intracellular concentrations, leading to multidrug resistance (MDR) in cancer and treatment failure. Interactions from the TME like hypoxic conditions induced by Reactive oxygen species (ROS), altered glucose metabolism (e.g. altered expression of enzyme like GLUT and upregulation of glycolytic pathways) and increased paracrine signaling of TAMs leads to an immunosuppressive environment, rendering immunotherapy ineffective.
2.3. The Role of Tumor Evolution in Driving Intratumoral Heterogeneity in Metastasis
Tumor cell heterogeneity evolves progressively as the tumor continues to grow, therefore tumor evolution plays a critical role in driving ITH. Several theories have emerged explaining how heterogenous cells arise within a tumor [34,35]. The clonal evolution theory, proposed by Peter Nowell in 1976, posits that mutations gradually accumulate within a single cancer cell that undergoes cell divisions to form distinct cell lineages [34]. Through natural selection, these subclones continue to acquire mutations, expand under selective pressures imposed by cancer therapy or eventually become extinct. The rare competent clones that survive the multiple pressures comprise of diverse subpopulations leading to ITH in primary tumor. As the tumor evolves, more mutations are acquired which can result in the selection of a few clones showing higher invasive ability and can potentially lead to metastasis. An example of this theory is indicated in a computational study where researchers identified a rare ancestral primary tumor subclone that was responsible for seeding metastasis in breast cancer patients. This subclone constituted only about 2% of the primary tumor cells but was detected in 29% metastatic breast cancer patients. Two significant mutations (PEAK1 K140Q and LRP5 A65V) acquired by this subclones were implicated in enhanced cell migration and metastatic potential compared to rest of the tumor cells [35].
Although these mutations were thought to have been responsible for survival of the subclones, this could likely be a random event rather than inherent fitness. This hypothesis could be further investigated using lineage tracing strategies, which allows for the tracking of the timing of dissemination of heterogeneous tumor cells. This hypothesis was put forward into two models called the early and late dissemination models. The early dissemination model predicts that cells from the primary tumor disseminate at a very early stage of tumor development or even before the clinical manifestation of the primary tumor. These DCCs remain dormant for extended periods, before migrating and colonizing the secondary sites. During the dormancy state, these cells could acquire mutations and continue to evolve, resulting in subpopulations exhibiting different genetic and phenotypic profiles, leading to ITH [14]. For instance, single-cell RNA sequencing of HER2-, HER2+ EL (early lungs) and HER2+LL (late lungs) DCCs revealed a transcription factor (ZFP281) driving early dissemination of cancer cells as well as inducing dormancy like state within a cluster of early DCCs [19]. On the other hand, the late dissemination model is the more widely accepted theory which suggests that DCCs will migrate once the tumor has attained significant size and invasive potential. The dissemination, therefore, happens at a later stage, where the tumor exhibits advanced neoplastic properties, giving rise to fully malignant late DCCs. These late DCCs acquire higher level of mutations and exhibit a more aggressive phenotype among metastatic patients [14]. An example of this theory was shown in a phylogenetic study that illustrated differences in genomic signatures between primary Osteosarcoma and matched pulmonary metastatic tumors in patients using multiregional whole genome sequencing. The metastatic tumors exhibited higher levels of germline mutations in DNA damage response genes (like MMR gene) as compared to the primary tumors. Phylogenetic analysis further revealed that these mutations occurred due to late dissemination of tumor cells, which drove a more aggressive phenotype in pulmonary metastatic cancer cell [36]. Therefore, the late dissemination model, for most instances suggests that late DCCs contribute to aggressive tumors.
The cancer stem cell (CSC) model is one of the most studied models which provides a paradigm for understanding ITH in metastatic cancer as well as therapeutic resistance. CSCs are a subset of cells within the tumor that are characterized by the ability to self-renew and differentiate into various cell types. This model suggests that tumors are hierarchically organized, with CSCs residing at the apex. As these cells differentiate, a small fraction of the CSCs can initiate and sustain tumor growth leading to distinct phenotypes [37]. CSCs was first reported by Lapidot et al. in acute myeloid leukemia, where a small group of CD34+CD38− tumor cells exhibited a phenotype similar to that of hematopoietic stem cells and showed a greater propensity towards tumor initiation. CSCs usually reside at the hypoxic core of the tumor in a quiescent state and are responsible for tumor recurrence or relapse post-therapy. Tabuchi et al. identified two distinct subclonal populations (sphere-like S and Leukemia-like LL clones) within CSCs in uterine endometrial carcinoma and classified them based on different proliferation rates and response to chemotherapy. Transcriptomic analysis (SAGE-seq) revealed that S clones are chemoresistant cells, less tumorigenic and show upregulated MAPK pathway, whereas L1 clones are highly tumorigenic and sensitive to chemotherapy [38]. Therefore, potential targets against the resistant CSCs can help in improving the efficacy of metastatic cancer therapeutic modalities.
2.4. Current Technologies and Computational Models to Study Heterogeneity in Metastasis
At present, identifying cells with varying metastatic potential and understanding the arrival of these resistant tumor cells, remains a major obstacle in cancer research. Moreover, recreating tumor models that mimic the physiological complexity of ITH in the context of the human body, has been an onerous task [39]. Nonetheless, some of the technical hurdles have been addressed through recent advancements in model systems and improvised technologies to investigate the dynamics of metastasis and therapeutic resistance. Some of the model systems that include PDX, genetically engineered mouse models (GEMM), bioprinting models, organoids, biomimetic tumor models and chip-on-chip models, that have also been modified to account for extrinsic factors in the TME. For instance, the traditional GEMM models failed to account for the Microsatellite instability subtype of gastric cancer. Through electroporation and the use of CRISPR-Cas9, researchers developed a new mouse model that accounted for all subtypes of metastatic gastric cancer. This new GEMM model called Electroporation-based Genetically engineered mice models (EPO GEMMs) are immunocompromised transgenic mice that spontaneously develop malignancies at the site of electroporation by inducing somatic mutations [40,41]. PDX is another model system in which tumor tissues from patients are implanted into immunocompromised mice [42,43]. The PDX model was used to study sequential treatment of PARP inhibitor olaparib and the chemotherapy drug oxaliplatin in metastatic colorectal cancer patients [44]. BEHAV3D is an example of a multispectral, 3D image-based platform which was used to track the separation of metabolome-sensing engineered T cells (TEG) of breast cancer into nine subpopulations, each exhibiting unique behavioral patterns (ranging from inactive to active mobility). Thus, the researchers were able to demonstrate nine unique phenotypes. This model can be used to devise a mode of action of cellular immunotherapy by predicting engineered T cell behavior within specific culture conditions [45]. In another study, a biomimetic model, ductal tumor-microenvironment-on-chip (dT-MOC), was designed to study EMT and the local invasion in Pancreatic Ductal Adenocarcinoma. This model comprises a microfluidic platform with a duct of murine genetically engineered pancreatic cancer cells embedded within a collagen matrix. The model demonstrated differential expression of biomarkers with eKIC cells expressing high levels of E-cadherin (Cdh1) and low levels of the EMT marker Snail. In contrast, mKIC cells exhibited high levels of Snail, low levels of Cdh1 and an exclusive mesenchymal phenotype. This E-cadherin suppression suggests that mesenchymal subtypes of cancer cells can have an effect on other epithelial subtypes and enhance the EMT of epithelial subtypes of cancer cells [39].
These model systems coupled with single cell profiling tehcnologies was used to first study ITH at genomic, transcriptomic, epigenomic and proteomic levels in cellular populations [41,46]. By amplifying DNA within individual cells, single-cell genomic techniques like multiple displacement amplification (MDA), multiple annealing and looping-based amplification cycles (MALBAC) and degenerate oligonucleotide-primed PCR (DOP-PCR), allowed in studying the de novo germline mutations and somatic mutations within cancer cells. In one study, topographic single cell sequencing (TSCS) was used to analyse genomic copy number of tumors to validate multiclonal invasion model of breast cancer evolution (from ductal carcinoma in situ to invasive ductal carcinoma). Single-cell DNA sequencing (scDNA-seq) was also used to understand the clonal evolution leading to the acquisition of resistance to FLT3 inhibitors in acute myeloid leukemia (AML) and accumulation of somatic mutations in B lymphocytes of B cell cancers [31,47].
Single cell transcriptomic sequencing measures transcript levels within a cell (scRNA-seq) or a variety of cells (bulk RNA-seq), and are by far the most widely used conventional methods to study ITH at transcriptomic level. scRNA-seq techniques like Smart-seq3, Smart-seq24, Quartz-Seq5, RamDa-seq7 and CEL-seq6 have been developed to measure mRNA from a single cell in a stable environment. Protocols to enable efficient handling of RNA from millions of single cells have been developed using microdroplet technology (Drop-Seq8 and DroNc-seq9) and microwell technology (Nx1-seq11 and Seq-Well12). Using scRNA-seq, Chung et al. also studied heterogeneity in Tumor-infiltrating lymphocytes among breast cancer using [48]. Tirosh et al. used scRNA-seq to quantify the T cell exhaustion programs within CD45+ and CD45− in each patient suffering from melanoma, which can be used to improve immunotherapy strategies [49]. Kashima et al. conducted scRNA-seq of lung cancer cell lines stimulated by receptor tyrosine kinase inhibitors and observed different transcriptional responses to the drug among sensitive and insensitive cells, thereby highlighting early resistance responses like dormancy [46]. scRNA-seq has also been used to identify progenitor cancer cell populations obtained from primary mammary and lung tumor and metastatic cells in PDX. Furthermore, this technology enabled characterization of resistant genotypes that existed prior to neoadjuvant chemotherapy and transcriptional programs that emerged after chemotherapy in TNBC tumors [41]. Single-cell cellular indexing of transcriptomes and epitopes by sequencing (scCITE-seq) is another technique that was used to identify different PD-L1/PD-L2+ macrophage populations and to study their interactions with stromal immune microenvironment in breast cancer metastasis. Moreover, this technique helped in showing differential expression of protein surface markers (CD117, CD49F and CD81) in primary and metastatic melanoma cells [50,51].
Recently, single-cell epigenomic sequencing technologies have enabled us to study epigenetic modifications like DNA methylation, chromatin accessibility, histone modification and chromatin structure, which play a critical role in ITH. For instance, single-cell chromatin immunoprecipitation followed by sequencing (scChIP-seq) has been designed which uses a droplet microfluidics-based procedure known as Drop-ChIP55, allowing identification of subpopulations based on unique chromatin signatures of pluripotency and histone modifications within Mouse embryonic stem cells (mEScs) and Mouse embryonic fibroblasts (mEFs). It was found that MEFs are lineage committed cells with a constrained chromatin state whereas MESc chromatin has a more accessible and plastic state [46]. Grosselin et al. used scChIP-seq to study the chromatin modulation contributing to ITH in PDX of breast cancer. The PDX treated with chemotherapy, resulted in chemosensitive and chemoresistant cell populations and both of which shared a common chromatin signature called H3K27me3 landscapes [52].
Single-cell proteomic sequencing technologies like CyToF, a method based on mass cytometry, was used to analyze the expression of surface and intracellular proteins by using antibodies tagged with metal labels [53]. Multiple studies have used a combination of aforementioned techniques and designed computational models to analyse the overall data. In combination with scRNA-seq, scATATC seq uncovered the intra cell- line heterogeneity at transcriptomic and epigenetic level in cancer cell lines among different tissue origins on account of CNVs, transcription factors like IRF1, IRF 2, STAT2 and extrachromosomal circular DNA [50,54]. The data obtained from scDNAseq was used to formulate a probability mixture model called sc clone to identify five distinct sub-clones in metastatic colorectal cancer [54].
Although single cell profiling technologies like scRNA seq, scATAC-seq, scCITE-seq allow characterization of tumor-specific cell types, as well enables studying the genetic, epigenetic, transcriptomic and proteomic modifications at the single-cell level, they do not account for physical organization of cells and excludes the cell specific interactions with extracellular matrix. To overcome these limitations, spatial profiling technologies have been developed that are capable of profiling gene expression levels in situ and analyzing the sub-clonal populations and their interactions within TME [50,55,56,57]. In recent years, both the technologies are used simultaneously (collectively termed as multiomics technology) to monitor critical stages of metastatic setting, including clonal and populations of primary tumor cells which escaped and invaded the local/distant parts of the body and tracing evolutionary origins of the sub-clonal populations [58]. The spatial profiling technologies can be broadly classified into Spatial Transcriptomic technologies and Spatial Proteomic analyses [59]. Spatial Transcriptomic profiling technologies use transcripts to monitor heterogeneity on spatial scale and this can be done using Imaging-based methods and In situ capture-based methods. Imaging-based methods like Multiplex imaging technologies capture high spatial resolution of thousands of images and can resolve the precise position of individual RNA molecules in tissue sections. In one study, it was used to track intratumoral differences in immune cell compositions in metastatic prostrate cancer [25]. MALDI-MS (matrix-assisted laser desorption ionization mass spectrometry imaging) was used to study metabolic heterogeneity within tumor microenvironment of breast cancer. MIBI-TOF (multiplexed ion beam imaging by time-of-flight) was used to classify TNBC into compartmentalized (exclusively tumor cells or immune cells, characterised by PD-1 expression on CD4+ T cells and PD-L1 expression on myeloid derived cells in the tumor–immune cell boarder) or mixed tumors (spatial mixing of immune cells and tumor cells, which had PD-L1 expression on tumor cells and PD-1 expression on CD8+ T cells) [27]. MERFISH platform enabled identification of two different populations of RNA (cell cycle-dependent and independent) inside the same tumor cell [60]. CosMx was used to compare TAMs in primary tumors and metastases ( breast cancer, colorectal cancer). In another study, it helped in identification of 10 unique tumor microenvironments within non-small cell lung and breast cancer, providing strong evidence of spatial heterogeneity inside a single cell. On the other hand, in situ capture-based technologies sequence the entire protein-coding transcriptome, thereby enabling us to have a better understanding of cellular mechanisms. Using 10X Visium, a spatial transcriptomics platform, researchers identified different expressions of ERBB2 biomarker between two clusters of metastatic breast cancer separated by non-neighbouring UMAP region [61]. NanoString GeoMx Digital Spatial Profiler was used to study intratumoral heterogeneity and expression of numerous biomarkers in breast tumor samples treated with Her-2-targeted therapy and which tested positive for CD4+ biomarker [62].
Multiplexed proteomic detection techniques allow us to study protein interaction present in the tumor with the TME using fluorophore or metal tags. Fluorophore-based methods include InSituPlex, MACSima and co-detection by indexing (CODEX) use primary antibodies labelled with DNA barcodes to reduce the time associated with multiplexed imaging, enabling the detection of more than 50 proteins. Metal-based imaging techniques include mass cytometry and multiplex ion beam imaging use detection of metal isotope-labelled antibodies with laser desorption mass spectrometry, enabling the measurement of proteins without interference [63].
Recent advances in bioinformatic modeling have revolutionized our capacity to delineate ITH by combining multiomic datasets (e.g., single-nucleotide variants, copy-number alterations, and spatial transcriptomics) to infer clonal phylogenies and detect phenotype-specific biomarkers [64]. For instance, the Multiomic and Multiscale Analysis (MOMA) platform applied to IDH-mutant astrocytomas identified unique malignant clones with specific spatial distributions and transcriptional profiles, while accounting for stromal contamination in bulk and single-nucleus sequencing data [65]. Likewise, scRNA-seq of MMTV-PyMT mouse metastatic breast cancer cell lines revealed tissue-specific patterns of gene expression, i.e., activated EMT markers (AEBP1, ITGB5 , FN1) within lung metastases, reflective of clinical impressions of phenotypic plasticity [66]. These computational approaches are now pivotal in linking transcriptional heterogeneity to functional outcomes, such as organ-specific metastasis and drug resistance. For example, clonal tracking in leukemia PDX models combined with scRNA-seq demonstrated that subpopulations with distinct gene expression profiles exhibit predictable, therapy-specific resistance patterns [67]. Nevertheless, reconciling spatial and temporal heterogeneity, especially when modeling dynamic relationships between tumor cells and immune-stromal niches that promote clonal evolution, is still challenging [41,68]. Emerging strategies, including imaging mass cytometry and 3D organoid-based spatial transcriptomics, are addressing these gaps by capturing microniches within tumors that correlate with clinical outcomes.
Machine learning (ML) has emerged as a cornerstone in deciphering metastasis-specific heterogeneity, specifically in predicting subtype-specific behaviors and therapeutic vulnerabilities. For instance, a recent study applied ML to preclinical brain metastasis models, revealing distinct electrophysiological signatures across breast, melanoma, and lung cancer subtypes. Principal component analysis of local field potential oscillations identified delta/theta band disruptions as universal metastasis markers, while alpha/gamma oscillations differentiated breast (E0771-BrM) and melanoma (B16/F10-BrM) subtypes. Decision Tree classifiers achieved 77% accuracy in predicting metastasis presence and subtype from spectral data [69]. These results highlight the potential for ML to noninvasively diagnose metastasis subtypes and inform personalized treatment strategies. Complementing this, computational frameworks like Cellular Potts Model and ordinary or partial differential equation-based approaches are elucidating the multi-scale dynamics in a tumor microenvironment, including the tumor-immune interactions. These models can be deterministic or stochastic in nature and provide insights into the complex interactions among tumor growth, invasion and therapeutic response [70,71,72,73,74,75]. For instance, the Cellular Potts Model allows researchers to simulate tumor behavior and interactions with immune cells, enhancing our understanding of how tumors evolve and respond to treatments [76,77]. Additionally, mathematical modeling has been instrumental in studying the interactions between various immune cells and tumors, highlighting the importance of the immune system in controlling cancer progression [75,78]. Meanwhile, in silico optimization algorithms are redefining combination therapy design: RNAi-based modeling of heterogeneous tumors demonstrated that optimal drug regimens often exclude agents targeting dominant subpopulations, instead prioritizing combinations that minimize residual fitness across all clones. This approach, validated in lymphoma PDX models, enhanced survival by repressing emergent resistance mechanisms [79]. Together, these advances show how bioinformatic modeling bridges molecular heterogeneity to clinical outcomes, offering a roadmap for overcoming resistance to therapy.
Single cell sequencing and spatial transcriptomics have transformed research on metastatic niches to offer high-resolution information on cellular heterogeneity as well as on microenvironmental interactions. Approaches like scRNA-seq and spatial transcriptomics enable one to chart the gene expression landscape at single-cell resolution without disrupting spatial context and thereby determine differentiated cellular niches within metastasis sites. For example, the computational framework scNiche integrates multi-view features of cells, including molecular profiles and neighboring cellular compositions, to identify and characterize cell niches in tissues. Applied to triple-negative breast cancer, scNiche revealed patient-specific niches with distinct phenotypic characteristics, such as inflamed macrophages co-localizing with stromal cells, which drive tumor progression and therapy resistance [80]. Similarly, spatial transcriptomics has been employed in colorectal cancer to dissect the tumor microenvironment at subcellular resolution, uncovering cancer-associated fibroblast subtypes that modulate immune evasion through TGF-β signaling pathways[81]. These technologies have also been utilized in neuroblastoma, where single-cell transcriptomic and epigenomic profiling of bone marrow aspirates demonstrated subtype-specific cellular plasticity and tumor-promoting interactions with macrophages exhibiting both M1 and M2 features [82]. Even with these advancements, the issues of high expense, complexity of data, and low resolution still remain. New approaches such as MERFISH and Stereo-seq aim to overcome these limitations by using image-based methods with spatially resolved transcriptomics in order to attain historically unprecedented resolutions in mapping metastatic niches[83].
In silico approaches are pivotal in identifying biomarkers that can predict drug resistance and metastatic potential, leveraging computational tools and multiomic datasets to uncover molecular mechanisms. For example, high-throughput pharmacogenomic screens combined with CRISPR data have identified intrinsic resistance biomarkers in cancer cell lines. A study analyzing the Genomics of Drug Sensitivity in Cancer (GDSC) and Cancer Therapeutics Response Portal (CTRP) datasets highlighted genetic alterations such as the EGFR T790M mutation and PTEN loss in lung adenocarcinoma cell lines resistant to EGFR inhibitors, providing actionable insights into therapy optimization [84] . Similarly, integrating gene expression profiles with drug sensitivity data has revealed mechanisms of response to genotoxic agents like cisplatin and doxorubicin. Baseline gene expression analysis identified differentially expressed genes such as TOP2A and XRCC1, which are associated with DNA damage repair pathways and intrinsic resistance [85]. Network-based approaches, such as protein-protein interaction (PPI) modeling, have also been employed to predict resistance pathways. In colorectal cancer, protein-protein interaction network analysis identified KRAS-mediated signaling as a central node contributing to resistance against anti-EGFR therapies, highlighting its role in tumor progression and therapeutic resistance [86]. Machine learning algorithms, such as Random Forests and Support Vector Machines, have been applied to multiomic datasets to predict therapy responses, enabling integrative analyses of transcriptomic profiles and drug sensitivity [87,88]. These computational frameworks not only reveal actionable biomarkers but also enable the formulation of combination therapies to evade resistance. For example, biomarker-driven drug combination strategies have demonstrated that targeting serine hydrolase enzymes like MGLL can sensitize resistant cancer cells to histone lysine demethylase inhibitors such as GSK-J4 [89]. Boolean network models, which simulate biomolecular regulation, have been employed to decode CRC tumorigenesis and evaluate therapeutic interventions. For example, the in silico Drosophila Patient Model (DPM) integrates patient-specific mutation data with RNA-seq gene expression profiles to predict cell fate outcomes and validate cytotoxicity of FDA-approved drugs. This model identified synergistic drug combinations such as paclitaxel-regorafenib and docetaxel-bortezomib, which demonstrated a 100% increase in apoptosis and a 100% decrease in proliferation in preclinical CRC studies [90].
Despite these advancements, challenges remain in validating biomarkers against heterogeneous tumor microenvironments, scalability and validation across diverse patients’ cohorts, remain significant barriers to translating computational predictions into clinical settings. Another significant barrier is the integration of multi-omics datasets, often generated using different platforms and methodologies. This heterogeneity in data collection makes it challenging to establish coherent computational frameworks that can well represent tumor evolution and response to therapy. Furthermore, cross-validation of predictive models in diverse patient cohorts is still a bottleneck. Tumor heterogeneity differs not only among cancer types but also among patients and generalizing preclinical model results from models such as patient-derived xenografts (PDX) to clinical settings is therefore challenging. Addressing these challenges requires interdisciplinary collaboration between computational scientists, molecular biologists, and clinicians to develop robust models that can integrate diverse datasets while accounting for dynamic tumor behavior.
The future of bioinformatic modeling lies in integrating emerging technologies with advanced computational frameworks to address the complexities of tumor heterogeneity. One promising avenue is the development of bioprinted tumor models that combine organoids with extracellular matrix (ECM) components to mimic the genetic, histological, and functional aspects of cancer heterogeneity. These models allow researchers to replicate cell growth, differentiation, and proliferation in physiologically relevant environments, providing a robust platform for studying tumor microenvironment dynamics and drug resistance mechanisms [41,91]. For example, bioprinted models incorporating ECM stiffness and architecture have successfully reproduced immune cell polarization and stromal interactions, paving the way for personalized therapeutic strategies. Another innovative approach involves organ-on-a-chip systems, which integrate 3D structures such as microvasculature and chemical stimuli to recreate the dynamic heterogeneity of the tumor microenvironment. These systems enable real-time monitoring of immune cell recruitment and phenotypic changes, offering valuable insights into tumor-immune interactions. While these technologies are still in their infancy, their potential to enhance preclinical studies and improve therapeutic precision is immense. Advances in multiscale modeling and machine learning are poised to revolutionize bioinformatic approaches to tumor heterogeneity. Multiscale models simulate cancer progression by integrating molecular mechanisms with cellular and tissue-level dynamics, providing unprecedented insights into clonal evolution and drug response [41]. For example, approximate Bayesian computation (ABC) has been used to fit model parameters based on omics data, enabling predictions of tumor behavior under various therapeutic pressures. Concurrently, machine learning algorithms such as generative adversarial networks (GANs) complement these models by creating robust predictive frameworks that incorporate physicochemical constraints [92]. By combining these computational tools with experimental data from patient-derived models, researchers can create digital twins that simulate individual tumor dynamics, unlocking new possibilities for personalized medicine.

Table 1.
Hallmarks and limitations of modelling systems and technologies used to study intratumoral heterogeneity.
Table 1.
Hallmarks and limitations of modelling systems and technologies used to study intratumoral heterogeneity.
| Name of the model | Hallmarks | Limitations | References |
|---|---|---|---|
| Electroporation based Genetically engineered mice models (EPO GEMMs) | They are immunocompromised transgenic mice that spontaneously develop malignancies at the site of electroporation by introducing somatic mutations | Cannot accumulate as many somatic mutations as organoid models Cannot provide the cell of origin for tumor development |
[40,41] |
| Organoids | 3D models best used to study spatio-temporal dynamics. Built using stem cells (ASCs, iPSCs, ESC) from primary tumor and patient derived organoids (PDOs) | Extremely challenging to maintain due to lack of standardization protocol | [45] |
| Lineage tracing | Provides critical information during organ development and insights into the molecular mechanisms of cancer origin. Helps in real time dynamic tracing of CSCs. |
Unable to capture the molecular phenotype of each profiled cell | [50] |
| Bioprinting models | Used in combination with organoids to mimic tumor microenvironment, spatial distribution and helps in understanding stromal cell intravasation. | Absence of vasculature network | [95] |
| Biomimetic Tumoroids | Recreates the spatial distribution of nutrients and oxygen, to demonstrate cancer cell heterogeneity and the way it affects the vascular network formation | Lack of immune surveillance | [39] |
| On-a-chip models | Allows replication of dynamic culture systems to mimic the heterogeneity of the tumor microenvironment (microvasculature, immune cells, physicochemical TME) | Lack of extra cellular matrix expression | [52] |
| Single cell RNA sequencing (scRNA-seq) | Conversion of RNA into cDNA using non-probe RNA-seq technology, This technique uses microfluidics (Drop-seq) and inDROP system to generate droplets, which encapsulates microbeads with barcodes for reverse transcription amplification. The barcodes present are attached to individual gene, which can also enable tracing the origin of each gene. | Insufficient to detect rare subpopulations within tumor mass, disseminated tumor cells and extracellular matrix Prone to allelic dropout and excludes ECM False-positive errors associated with massive amplification of DNA Developing computational algorithms to analyse data on massive scale is a huge challenge Difficult to use in therapy-naïve primary lesions |
[48] |
| Single-cell assay for transposase accessible chromatin by sequencing (scATAC-seq) | Uses transcription factors and cis/trans regulatory elements for studying epigenetic modifications. Enables simultaneous profiling of accessible chromatin and protein levels, using transposase |
Exclusively binary data output Spatial mapping from closed to open chromatin is more difficult. Fails to detect rare or transient cell states or regulatory elements. |
[59] |
| Single-cell cellular indexing of transcriptomes and epitopes by sequencing (scCITE-seq) | Antibody panels are tested for detecting epitopes of interest, thus allowing simultaneous study of transcriptomic expression and cell surface protein marker. | Sensitive to enzymatic digestion due to loss of surface epitopes Difficult to perform antibody panel testing for developing the epitopes due to limited sample size |
[50] |
| Single-cell Clone (ScClone) | Employs probability mixture model from scDNA-seq data , by characterizing single cell into distinct subclones | Assumes genotypic errors are uniformly distributed leading to amplification bias. Does not explicitly model doublet events so its performance quality can get degraded |
[50] |
| Multiplex imaging | Allows spatial visualization and quantification of cell populations within metastatic tumors Generates multiple images of high-parameter protein biomarkers using an in-situ polymerization-based indexing procedure in conjugation with mass spectrometry, fluorescence-based microscopy and antibody-targeted sequencing |
Standardization of methods is needed Errors related to visual inspection due to huge dataset size Limited resolution to visualize overlapping cell fragments and irregularly shaped cell types (e.g. macrophages) |
[25] |
| NanoString GeoMx Digital Spatial Profiler (DSP) |
A novel high-plex, non-destructive protein and RNA profiling technique used to detect tumor heterogeneity in frozen or formalin fixed paraffin embedded (FFPE) tumor samples. It quantifies the protein or RNA by counting unique indexing oligonucleotides, thereby allowing large number of biomarkers to be studied on spatial temporal scale |
Does not provide single-cell resolution information at spatial level. | [62] |
| NanoString CosMxTM | An upgradation of GeomX DSP used to simultaneously study localization of RNA at cellular and subcellular level. | Limited resolution of very small sized cells and overlapping cells. | [61] |
| 10X Visium |
Uses oligonucleotides immobilised on special glass slide (visium) to locate mRNA from fixed tumor samples, which are sequenced using Next Generation Sequencing | Limited resolution of very small sized cells and overlapping cells. | [61] |
| MERFISH |
Multiplexed single-molecule imaging technology used to simultaneously analyse thousands of RNA on spatial scale. | Variability in resolution of the data ranging from few to hundreds spots (number of cells within a single spatial region) | [60] |
3. Discussion
Over the years, significant progress has been made in improving diagnosis and therapeutic regimens for metastatic cancer, yet the ultimate goal of achieving a cure for metastatic cancer remains elusive. One of the major factors hindering the progress is intratumor heterogeneity. Despite technological advances, identifying and studying heterogenous sub clones within tumor as well as interpreting humongous amounts of data on spatiotemporal scale is challenging for clinical researchers. The task of procurement and characterization of samples within different sites in metastatic tissue samples is daunting. This is currently addressed using multi region sampling strategy, rapid autopsy, warm autopsy and recently developed rapid postmortem tissue procurement program (after death). However, these protocols have failed to efficiently detect the small (7- 10 microns) aggressive disseminated tumor cells (DTCs), which contribute for most instances of metastatic relapse [55,58]. No protocol for collecting metastatic tumor tissues, such that it accounts for all the sub-clones of the tumor, has ever been formulated.
Besides the size, it is the uncertain state of activity and prolonged dormancy of DTCs, which makes it challenging to detect DTCs [58]. Therefore, detection of these sub-clonal populations within DTCs will provide insights into the role of this heterogenous population and enable timely treatment [55,58]. In a recent study, Bacon et al. [93] attempted to trace the heterogenous population of cells in metastatic breast cancer using the predictive biomarker PD-L1 using immunohistochemistry assay. However, this biomarker evaluation is based on patient’s early metastatic biopsy, rather than from a real-time metastatic site. Therefore, increasing frequency of biopsies to monitor real-time progression and change in heterogenous composition within metastatic sites is a proposition for enhanced therapeutic strategies [55]. In addition to this, limited availability of metastatic tumor samples is a major hindrance in studying metastatic ITH. Therefore, increasing access to research autopsies would help in standardizing and making clinical trials more conclusive. Although there have been advances in spatial profiling technologies, there is paucity in developing technologies to monitor the cancer cell cycle heterogeneity. Since the cell cycle phase can determine the response of cancer cells towards drug, understanding of the role of heterogeneous subpopulations with varying cell-cycle dynamics will facilitate in developing efficient treatment. Further studies using mathematical modelling, single cell sequencing and meta-omics analysis will help us further unfold the secrets of metastatic heterogeneity.
Besides technical challenges, establishing metastatic patient cohorts poses several challenges. Metastatic cancer patients are often in a compromised state of health, therefore approaching the patients at a critical stage remains a daunting task. Due to sheer exertion and side effects of ongoing therapies, they may struggle to comprehend the risks and benefits of participating in these cohorts and therefore unwilling to provide informed consent. Establishing a cohort that encompasses and equally represents geographical and demographic diverse populations is nearly impossible, in a metastatic setting [55]. Moreover, patient selection bias is a huge matter of concern as most metastatic patients fail to meet the inclusion criteria due to inconsistences and inaccuracies in their medical history details like medical issues prior to diagnosis of metastatic cancer, during clinical data collection process. Additionally, studying ITH within different metastatic sites and throughout metastatic disease progression adds another layer of complexity [94].
Besides the technical challenges, several region-specific obstacles continue to hinder progress in metastatic cancer research. In a country like India, with limited resources, the situation is further complicated owing to the nation's extensive and heterogeneous population. The enormous genetic and cultural heterogeneity among regions hampers the establishment of homogenous cohorts, while inequalities in healthcare facilities between urban and rural areas further hinder recruitment. A significant number of patients, especially in rural areas, have limited access to tertiary care facilities, and those who do may be uninformed about clinical research or reluctant to engage due to apprehensions around privacy or exploitation. The lack of standardized electronic medical records and discrepancies in diagnostic and therapeutic techniques among institutions lead to inconsistencies in data collection and quality. These hurdles are exacerbated by practical and financial limitations, such as insufficient funding for biobanking and obstacles in the preservation and transportation of samples from remote locations. Resolving these difficulties necessitates a unified endeavor to enhance awareness, infrastructure, and standardization throughout the healthcare and research ecosystem.
India has several clinical resources that can be leveraged to address metastatic heterogeneity despite these challenges. Regional cancer centers and tertiary institutes, such as Tata Memorial Hospital and AIIMS, provide access to large and diverse patient pools, making them key hubs for metastatic studies. Emerging biobanking initiatives like the National Cancer Tissue Biobank (NCTB) are facilitating the availability of high-quality biospecimens for research. Collaborative networks such as the National Cancer Grid promote data sharing and multicenter studies, helping to bridge regional disparities. Precision medicine programs focusing on genomic and transcriptomic profiling are becoming more prevalent, offering valuable insights into tumor heterogeneity and metastasis. Additionally, advancements in imaging modalities, such as PET-CT and MRI, combined with molecular diagnostics, enhance the ability to study real-time metastatic progression. Platforms like the Clinical Trials Registry of India (CTRI) further support clinical trial coordination, although challenges in patient recruitment remain. These resources, when utilized effectively, can significantly advance the understanding of metastatic heterogeneity in Indian populations.
In conclusion, the review highlights the role of ITH in metastasis and drug resistance. Addressing the molecular, technical and clinical challenges to study ITH in metastatic cancer patients would require a constant, multidisciplinary effort is to unravel the complexities of metastatic heterogeneity and enable efficient therapeutic interventions for diverse patient populations.
Author Contributions
RN, JSP and MKJ contributed to the study’s conception and design. NSB, JS, SS, RA and JSP conducted the literature review and drafted the initial manuscript. JS designed figures for the review. RN, JSP and MKJ supervised the manuscript and polished the language. The authors read and approved the final manuscript.
Funding
The authors declare that financial support was received for the research, and publication of this article. RN was supported by CHG intramural funding. MKJ and SS were supported by Param Hansa Philanthropies.
Institutional Review Board Statement
No datasets were generated or analyzed during the current study.
Data Availability Statement
No datasets were generated or analyzed during the current study.
Conflicts of Interest
The authors declare no competing interests.
Abbreviations
| TME | tumor microenvironment |
| ITH | intratumoral heterogeneity |
| CAF | cancer-associated fibroblast |
| TAM | tumor-associated macrophages |
| EMT | epithelial-mesenchymal transition |
| CRC | colorectal cancer |
| scRNA-seq | single-cell RNA sequencing |
| FACS | fluorescence activated cell sorting |
| 2Do | 2-dimensional patient organoids |
| PDAC | pancreatic ductal adenocarcinoma |
| LP | primary lung adenocarcinoma |
| BM | brain metastasis |
| SCNA | somatic copy number alteration |
| IFN-γ | Interferon gamma |
| HCC | hepatocellular carcinoma |
| DCC | disseminated cancer cells |
| PDX | patient-derived xenograft |
| PSA | prostate specific antigen |
| MCM3 | minichromosome maintenance protein 3 |
| MCT1 | Monocarboxylate transporter 1 |
| ROS | Reactive oxygen species |
| TNBC | Triple-negative breast cancer |
| CNV | Copy number variations |
| AR | androgen receptor |
| Macc1 | Metastasis associated colon cancer 1 |
| ABC | ATP-binding cassette |
| MDR | multidrug resistance |
| CSC | cancer stem cell |
| GEMM | Genetically engineered mouse model |
| EPO GEMM | Electroporation-based Genetically engineered mice model |
| dT-MOC | ductal tumor-microenvironment-on-chip |
| MDA | multiple displacement amplification |
| MALBAC | multiple annealing and looping-based amplification cycles |
| DOP-PCR | degenerate oligonucleotide-primed PCR |
| TSCS | topographic single cell sequencing |
| AML | acute myeloid leukemia |
| scCITE-seq | Single-cell cellular indexing of transcriptomes and epitopes by sequencing |
| scChIP-seq | single-cell chromatin immunoprecipitation followed by sequencing |
| mESc | Mouse embryonic stem cells |
| mEF | Mouse embryonic fibroblasts |
| MALDI-MS | matrix-assisted laser desorption ionization mass spectrometry imaging |
| MIBI-TOF | multiplexed ion beam imaging by time-of-flight |
| CODEX | co-detection by indexing |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| CTRP | Cancer Therapeutics Response Portal |
| DPM | Drosophila Patient Model |
| ECM | extracellular matrix |
| ABC | approximate Bayesian computation |
| GAN | generative adversarial networks |
| DTC | Disseminated tumor cells |
| NCTB | National Cancer Tissue Biobank |
| CTRI | Clinical Trials Registry of India |
References
- Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, Jemal A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2024;74(3):229–63. [CrossRef]
- Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Sig Transduct Target Ther. 2020 Mar 12;5(1):1–17.
- Mani K, Salgaonkar BB, Das D, Bragança JM. Community solar salt production in Goa, India. Aquat Biosyst. 2012 Dec 1;8(1):30. [CrossRef]
- Shi X, Wang X, Yao W, Shi D, Shao X, Lu Z, Chai Y, Song J, Tang W, Wang X. Mechanism insights and therapeutic intervention of tumor metastasis: latest developments and perspectives. Sig Transduct Target Ther. 2024 Aug 2;9(1):1–46. [CrossRef]
- Liu K, Han H, Xiong K, Zhai S, Yang X, Yu X, Chen B, Liu M, Dong Q, Meng H, Gu Y. Single-cell landscape of intratumoral heterogeneity and tumor microenvironment remolding in pre-nodal metastases of breast cancer. Journal of Translational Medicine. 2024 Aug 29;22(1):804. [CrossRef]
- El-Sayes N, Vito A, Mossman K. Tumor Heterogeneity: A Great Barrier in the Age of Cancer Immunotherapy. Cancers. 2021 Jan;13(4):806. [CrossRef]
- Sun X xiao, Yu Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol Sin. 2015 Oct;36(10):1219–27.
- Li D, Xia L, Huang P, Wang Z, Guo Q, Huang C, Leng W, Qin S. Heterogeneity and plasticity of epithelial–mesenchymal transition (EMT) in cancer metastasis: Focusing on partial EMT and regulatory mechanisms. Cell Proliferation. 2023;56(6):e13423. [CrossRef]
- Brown MS, Abdollahi B, Wilkins OM, Lu H, Chakraborty P, Ognjenovic NB, Muller KE, Jolly MK, Christensen BC, Hassanpour S, Pattabiraman DR. Phenotypic heterogeneity driven by plasticity of the intermediate EMT state governs disease progression and metastasis in breast cancer [Internet]. bioRxiv; 2022 [cited 2025 Mar 14]. p. 2021.03.17.434993. Available from: https://www.biorxiv.org/content/10.1101/2021.03.17.434993v2.
- Ramón y Cajal S, Sesé M, Capdevila C, Aasen T, De Mattos-Arruda L, Diaz-Cano SJ, Hernández-Losa J, Castellví J. Clinical implications of intratumor heterogeneity: challenges and opportunities. J Mol Med. 2020 Feb 1;98(2):161–77. [CrossRef]
- Wang X, Bai H, Zhang J, Wang Z, Duan J, Cai H, Cao Z, Lin Q, Ding X, Sun Y, Zhang W, Xu X, Chen H, Zhang D, Feng X, Wan J, Zhang J, He J, Wang J. Genetic Intratumor Heterogeneity Remodels the Immune Microenvironment and Induces Immune Evasion in Brain Metastasis of Lung Cancer. Journal of Thoracic Oncology. 2024 Feb 1;19(2):252–72. [CrossRef]
- Fujino S, Miyoshi N, Ito A, Hayashi R, Yasui M, Matsuda C, Ohue M, Horie M, Yachida S, Koseki J, Shimamura T, Hata T, Ogino T, Takahashi H, Uemura M, Mizushima T, Doki Y, Eguchi H. Metastases and treatment-resistant lineages in patient-derived cancer cells of colorectal cancer. Commun Biol. 2023 Nov 24;6(1):1–14. [CrossRef]
- Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021 Feb;27(2):212–24. [CrossRef]
- Pasha N, Turner NC. Understanding and overcoming tumor heterogeneity in metastatic breast cancer treatment. Nat Cancer. 2021 Jul;2(7):680–92.
- Peng Z, Ye M, Ding H, Feng Z, Hu K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J Transl Med. 2022 Jul 6;20(1):302. [CrossRef]
- Hapach LA, Carey SP, Schwager SC, Taufalele PV, Wang W, Mosier JA, Ortiz-Otero N, McArdle TJ, Goldblatt ZE, Lampi MC, Bordeleau F, Marshall JR, Richardson IM, Li J, King MR, Reinhart-King CA. Phenotypic Heterogeneity and Metastasis of Breast Cancer Cells. Cancer Research. 2021 Jul 1;81(13):3649–63.
- Roper N, Gao S, Maity TK, Banday AR, Zhang X, Venugopalan A, Cultraro CM, Patidar R, Sindiri S, Brown AL, Goncearenco A, Panchenko AR, Biswas R, Thomas A, Rajan A, Carter CA, Kleiner DE, Hewitt SM, Khan J, Prokunina-Olsson L, Guha U. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019 Mar 5;26(10):2651-2666.e6.
- Zhai W, Lai H, Kaya NA, Chen J, Yang H, Lu B, Lim JQ, Ma S, Chew SC, Chua KP, Alvarez JJS, Chen PJ, Chang MM, Wu L, Goh BKP, Chung AYF, Chan CY, Cheow PC, Lee SY, Kam JH, Kow AWC, Ganpathi IS, Chanwat R, Thammasiri J, Yoong BK, Ong DBL, de Villa VH, Dela Cruz RD, Loh TJ, Wan WK, Zeng Z, Skanderup AJ, Pang YH, Madhavan K, Lim TKH, Bonney G, Leow WQ, Chew V, Dan YY, Tam WL, Toh HC, Foo RSY, Chow PKH. Dynamic phenotypic heterogeneity and the evolution of multiple RNA subtypes in hepatocellular carcinoma: the PLANET study. National Science Review. 2022 Mar 1;9(3):nwab192. [CrossRef]
- Nobre AR, Dalla E, Yang J, Huang X, Wullkopf L, Risson E, Razghandi P, Anton ML, Zheng W, Seoane JA, Curtis C, Kenigsberg E, Wang J, Aguirre-Ghiso JA. ZFP281 drives a mesenchymal-like dormancy program in early disseminated breast cancer cells that prevents metastatic outgrowth in the lung. Nat Cancer. 2022 Oct;3(10):1165–80.
- Winkler J, Tan W, Diadhiou CMM, McGinnis CS, Abbasi A, Hasnain S, Durney S, Atamaniuc E, Superville D, Awni L, Lee JV, Hinrichs JH, Wagner PS, Singh N, Hein MY, Borja M, Detweiler AM, Liu SY, Nanjaraj A, Sitarama V, Rugo HS, Neff N, Gartner ZJ, Pisco AO, Goga A, Darmanis S, Werb Z. Single-cell analysis of breast cancer metastasis reveals epithelial-mesenchymal plasticity signatures associated with poor outcomes. J Clin Invest [Internet]. 2024 Sep 3 [cited 2025 Mar 14];134(17). Available from: https://www.jci.org/articles/view/164227.
- Hua X, Zhao W, Pesatori AC, Consonni D, Caporaso NE, Zhang T, Zhu B, Wang M, Jones K, Hicks B, Song L, Sampson J, Wedge DC, Shi J, Landi MT. Genetic and epigenetic intratumor heterogeneity impacts prognosis of lung adenocarcinoma. Nat Commun. 2020 May 18;11(1):2459. [CrossRef]
- Iglesias-Gato D, Thysell E, Tyanova S, Crnalic S, Santos A, Lima TS, Geiger T, Cox J, Widmark A, Bergh A, Mann M, Flores-Morales A, Wikström P. The Proteome of Prostate Cancer Bone Metastasis Reveals Heterogeneity with Prognostic Implications. Clin Cancer Res. 2018 Nov 1;24(21):5433–44.
- Zou Y, Ye F, Kong Y, Hu X, Deng X, Xie J, Song C, Ou X, Wu S, Wu L, Xie Y, Tian W, Tang Y, Wong CW, Chen ZS, Xie X, Tang H. The Single-Cell Landscape of Intratumoral Heterogeneity and The Immunosuppressive Microenvironment in Liver and Brain Metastases of Breast Cancer. Advanced Science. 2023;10(5):2203699. [CrossRef]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, Kasitinon SY, Vandergriff T, Mathews TP, Zhao Z, Schadendorf D, DeBerardinis RJ, Morrison SJ. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature. 2020 Jan;577(7788):115–20. [CrossRef]
- Moosavi SH, Eide PW, Eilertsen IA, Brunsell TH, Berg KCG, Røsok BI, Brudvik KW, Bjørnbeth BA, Guren MG, Nesbakken A, Lothe RA, Sveen A. De novo transcriptomic subtyping of colorectal cancer liver metastases in the context of tumor heterogeneity. Genome Medicine. 2021 Sep 1;13(1):143. [CrossRef]
- Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. cdr. 2019 Jun 19;2(2):141–60.
- Maleki EH, Bahrami AR, Matin MM. Cancer cell cycle heterogeneity as a critical determinant of therapeutic resistance. Genes & Diseases. 2024 Jan 1;11(1):189–204. [CrossRef]
- Ryl T, Kuchen EE, Bell E, Shao C, Flórez AF, Mönke G, Gogolin S, Friedrich M, Lamprecht F, Westermann F, Höfer T. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. cels. 2017 Sep 27;5(3):237-250.e8. [CrossRef]
- Peng J, Sun BF, Chen CY, Zhou JY, Chen YS, Chen H, Liu L, Huang D, Jiang J, Cui GS, Yang Y, Wang W, Guo D, Dai M, Guo J, Zhang T, Liao Q, Liu Y, Zhao YL, Han DL, Zhao Y, Yang YG, Wu W. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019 Sep;29(9):725–38.
- Yang F, Wang Y, Li Q, Cao L, Sun Z, Jin J, Fang H, Zhu A, Li Y, Zhang W, Wang Y, Xie H, Gustafsson JÅ, Wang S, Guan X. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis. 2017 Sep 1;38(9):900–9. [CrossRef]
- McMahon CM, Ferng T, Canaani J, Wang ES, Morrissette JJD, Eastburn DJ, Pellegrino M, Durruthy-Durruthy R, Watt CD, Asthana S, Lasater EA, DeFilippis R, Peretz CAC, McGary LHF, Deihimi S, Logan AC, Luger SM, Shah NP, Carroll M, Smith CC, Perl AE. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discovery. 2019 Aug 1;9(8):1050–63.
- Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, Gulati R, Plymate SR, Zhou Z, Birditt B, Meredith R, Geiss G, Hoang M, Beechem J, Nelson PS. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun. 2021 Mar 3;12(1):1426.
- Thankamony AP, Ramkomuth S, Ramesh ST, Murali R, Chakraborty P, Karthikeyan N, Varghese BA, Jaikumar VS, Jolly MK, Swarbrick A, Nair R. Phenotypic heterogeneity drives differential disease outcome in a mouse model of triple negative breast cancer. Front Oncol. 2023;13:1230647. [CrossRef]
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976 Oct 1;194(4260):23–8.
- Lawrence-Paul MR, Pan T chi, Pant DK, Shih NNC, Chen Y, Belka GK, Feldman M, DeMichele A, Chodosh LA. Rare subclonal sequencing of breast cancers indicates putative metastatic driver mutations are predominately acquired after dissemination. Genome Medicine. 2024 Feb 6;16(1):26. [CrossRef]
- Wang D, Niu X, Wang Z, Song CL, Huang Z, Chen KN, Duan J, Bai H, Xu J, Zhao J, Wang Y, Zhuo M, Xie XS, Kang X, Tian Y, Cai L, Han JF, An T, Sun Y, Gao S, Zhao J, Ying J, Wang L, He J, Wang J. Multiregion Sequencing Reveals the Genetic Heterogeneity and Evolutionary History of Osteosarcoma and Matched Pulmonary Metastases. Cancer Res. 2019 Jan 1;79(1):7–20.
- Tu SM, Zhang M, Wood CG, Pisters LL. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers. 2021 Jan;13(16):4006. [CrossRef]
- Tabuchi Y, Hirohashi Y, Hashimoto S, Mariya T, Asano T, Ikeo K, Kuroda T, Mizuuchi M, Murai A, Uno S, Kawai N, Kubo T, Nakatsugawa M, Kanaseki T, Tsukahara T, Saito T, Torigoe T. Clonal analysis revealed functional heterogeneity in cancer stem-like cell phenotypes in uterine endometrioid adenocarcinoma. Exp Mol Pathol. 2019 Feb;106:78–88. [CrossRef]
- Bradney MJ, Venis SM, Yang Y, Konieczny SF, Han B. A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma. Small. 2020;16(10):1905500. [CrossRef]
- Leibold J, Tsanov KM, Amor C, Ho YJ, Sánchez-Rivera FJ, Feucht J, Baslan T, Chen HA, Tian S, Simon J, Wuest A, Wilkinson JE, Lowe SW. Somatic mouse models of gastric cancer reveal genotype-specific features of metastatic disease. Nat Cancer. 2024 Feb;5(2):315–29. [CrossRef]
- Proietto M, Crippa M, Damiani C, Pasquale V, Sacco E, Vanoni M, Gilardi M. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front Oncol [Internet]. 2023 Apr 28 [cited 2025 Mar 14];13. Available from: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1164535/full.
- Liu Y, Wu W, Cai C, Zhang H, Shen H, Han Y. Patient-derived xenograft models in cancer therapy: technologies and applications. Sig Transduct Target Ther. 2023 Apr 12;8(1):1–24. [CrossRef]
- Georgopoulou D, Callari M, Rueda OM, Shea A, Martin A, Giovannetti A, Qosaj F, Dariush A, Chin SF, Carnevalli LS, Provenzano E, Greenwood W, Lerda G, Esmaeilishirazifard E, O’Reilly M, Serra V, Bressan D, Gordon B. Mills, Ali HR, Cosulich SS, Hannon GJ, Bruna A, Caldas C. Landscapes of cellular phenotypic diversity in breast cancer xenografts and their impact on drug response. Nat Commun. 2021 Mar 31;12(1):1998. [CrossRef]
- Arena S, Corti G, Durinikova E, Montone M, Reilly NM, Russo M, Lorenzato A, Arcella P, Lazzari L, Rospo G, Pagani M, Cancelliere C, Negrino C, Isella C, Bartolini A, Cassingena A, Amatu A, Mauri G, Sartore-Bianchi A, Mittica G, Medico E, Marsoni S, Linnebacher M, Abrignani S, Siena S, Di Nicolantonio F, Bardelli A. A Subset of Colorectal Cancers with Cross-Sensitivity to Olaparib and Oxaliplatin. Clinical Cancer Research. 2020 Mar 13;26(6):1372–84.
- Dekkers JF, Alieva M, Cleven A, Keramati F, Wezenaar AKL, van Vliet EJ, Puschhof J, Brazda P, Johanna I, Meringa AD, Rebel HG, Buchholz MB, Barrera Román M, Zeeman AL, de Blank S, Fasci D, Geurts MH, Cornel AM, Driehuis E, Millen R, Straetemans T, Nicolasen MJT, Aarts-Riemens T, Ariese HCR, Johnson HR, van Ineveld RL, Karaiskaki F, Kopper O, Bar-Ephraim YE, Kretzschmar K, Eggermont AMM, Nierkens S, Wehrens EJ, Stunnenberg HG, Clevers H, Kuball J, Sebestyen Z, Rios AC. Uncovering the mode of action of engineered T cells in patient cancer organoids. Nat Biotechnol. 2023 Jan;41(1):60–9.
- Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med. 2020 Sep;52(9):1419–27. [CrossRef]
- Zhang L, Dong X, Lee M, Maslov AY, Wang T, Vijg J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proceedings of the National Academy of Sciences. 2019 Apr 30;116(18):9014–9.
- Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, Ryu HS, Kim S, Lee JE, Park YH, Kan Z, Han W, Park WY. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017 May 5;8(1):15081. [CrossRef]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, Fallahi-Sichani M, Dutton-Regester K, Lin JR, Cohen O, Shah P, Lu D, Genshaft AS, Hughes TK, Ziegler CGK, Kazer SW, Gaillard A, Kolb KE, Villani AC, Johannessen CM, Andreev AY, Van Allen EM, Bertagnolli M, Sorger PK, Sullivan RJ, Flaherty KT, Frederick DT, Jané-Valbuena J, Yoon CH, Rozenblatt-Rosen O, Shalek AK, Regev A, Garraway LA. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016 Apr 8;352(6282):189–96.
- Chen S, Jiang W, Du Y, Yang M, Pan Y, Li H, Cui M. Single-cell analysis technologies for cancer research: from tumor-specific single cell discovery to cancer therapy. Front Genet [Internet]. 2023 Oct 12 [cited 2025 Mar 14];14. Available from: https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.1276959/full.
- Lischetti U, Tastanova A, Singer F, Grob L, Carrara M, Cheng PF, Martínez Gómez JM, Sella F, Haunerdinger V, Beisel C, Levesque MP. Dynamic thresholding and tissue dissociation optimization for CITE-seq identifies differential surface protein abundance in metastatic melanoma. Commun Biol. 2023 Aug 10;6(1):1–16. [CrossRef]
- Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F, Frenoy O, Pousse Y, Reichen M, Woolfe A, Brenan C, Griffiths AD, Vallot C, Gérard A. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet. 2019 Jun;51(6):1060–6. [CrossRef]
- Chen J, Larsson L, Swarbrick A, Lundeberg J. Spatial landscapes of cancers: insights and opportunities. Nat Rev Clin Oncol. 2024 Sep;21(9):660–74. [CrossRef]
- Zhu Q, Zhao X, Zhang Y, Li Y, Liu S, Han J, Sun Z, Wang C, Deng D, Wang S, Tang Y, Huang Y, Jiang S, Tian C, Chen X, Yuan Y, Li Z, Yang T, Lai T, Liu Y, Yang W, Zou X, Zhang M, Cui H, Liu C, Jin X, Hu Y, Chen A, Xu X, Li G, Hou Y, Liu L, Liu S, Fang L, Chen W, Wu L. Single cell multi-omics reveal intra-cell-line heterogeneity across human cancer cell lines. Nat Commun. 2023 Dec 9;14(1):8170.
- Parker AL, Benguigui M, Fornetti J, Goddard E, Lucotti S, Insua-Rodríguez J, Wiegmans AP, Early Career Leadership Council of the Metastasis Research Society. Current challenges in metastasis research and future innovation for clinical translation. Clin Exp Metastasis. 2022 Apr 1;39(2):263–77. [CrossRef]
- van Dam S, Baars MJD, Vercoulen Y. Multiplex Tissue Imaging: Spatial Revelations in the Tumor Microenvironment. Cancers. 2022 Jan;14(13):3170.
- Yu Z, Du F, Song L. SCClone: Accurate Clustering of Tumor Single-Cell DNA Sequencing Data. Front Genet [Internet]. 2022 Jan 27 [cited 2025 Mar 14];13. Available from: https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2022.823941/full.
- Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. 2018 Dec;20(12):1349–60. [CrossRef]
- Cheng M, Jiang Y, Xu J, Mentis AFA, Wang S, Zheng H, Sahu SK, Liu L, Xu X. Spatially resolved transcriptomics: a comprehensive review of their technological advances, applications, and challenges. Journal of Genetics and Genomics. 2023 Sep 1;50(9):625–40. [CrossRef]
- Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci U S A. 2019 Sep 24;116(39):19490–9. [CrossRef]
- Andersson A, Larsson L, Stenbeck L, Salmén F, Ehinger A, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Borg Å, Frisén J, Engblom C, Lundeberg J. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat Commun. 2021 Oct 14;12(1):6012.
- Hernandez S, Lazcano R, Serrano A, Powell S, Kostousov L, Mehta J, Khan K, Lu W, Solis LM. Challenges and Opportunities for Immunoprofiling Using a Spatial High-Plex Technology: The NanoString GeoMx® Digital Spatial Profiler. Front Oncol [Internet]. 2022 Jun 29 [cited 2025 Mar 14];12. Available from: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.890410/full. [CrossRef]
- Semba T, Ishimoto T. Spatial analysis by current multiplexed imaging technologies for the molecular characterisation of cancer tissues. Br J Cancer. 2024 Dec;131(11):1737–47.
- Blighe K, Kenny L, Patel N, Guttery DS, Page K, Gronau JH, Golshani C, Stebbing J, Coombes RC, Shaw JA. Whole Genome Sequence Analysis Suggests Intratumoral Heterogeneity in Dissemination of Breast Cancer to Lymph Nodes. PLoS One. 2014 Dec 29;9(12):e115346. [CrossRef]
- Schupp PG, Shelton SJ, Brody DJ, Eliscu R, Johnson BE, Mazor T, Kelley KW, Potts MB, McDermott MW, Huang EJ, Lim DA, Pieper RO, Berger MS, Costello JF, Phillips JJ, Oldham MC. Deconstructing Intratumoral Heterogeneity through Multiomic and Multiscale Analysis of Serial Sections. Cancers. 2024 Jan;16(13):2429. [CrossRef]
- Ionkina AA, Balderrama-Gutierrez G, Ibanez KJ, Phan SHD, Cortez AN, Mortazavi A, Prescher JA. Transcriptome analysis of heterogeneity in mouse model of metastatic breast cancer. Breast Cancer Research. 2021 Sep 27;23(1):93.
- Contreras-Trujillo H, Eerdeng J, Akre S, Jiang D, Contreras J, Gala B, Vergel-Rodriguez MC, Lee Y, Jorapur A, Andreasian A, Harton L, Bramlett CS, Nogalska A, Xiao G, Lee JW, Chan LN, Müschen M, Merchant AA, Lu R. Deciphering intratumoral heterogeneity using integrated clonal tracking and single-cell transcriptome analyses. Nat Commun. 2021 Nov 11;12:6522. [CrossRef]
- Xulu KR, Nweke EE, Augustine TN. Delineating intra-tumoral heterogeneity and tumor evolution in breast cancer using precision-based approaches. Front Genet [Internet]. 2023 Aug 17 [cited 2025 Apr 6];14. Available from: https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2023.1087432/full. [CrossRef]
- Sanchez-Aguilera A, Masmudi-Martín M, Navas-Olive A, Baena P, Hernández-Oliver C, Priego N, Cordón-Barris L, Alvaro-Espinosa L, García S, Martínez S, Lafarga M, Lin MZ, Al-Shahrour F, Menendez de la Prida L, Valiente M. Machine learning identifies experimental brain metastasis subtypes based on their influence on neural circuits. Cancer Cell. 2023 Sep 11;41(9):1637-1649.e11.
- Prasanna CVS, Jolly MK, Bhat R. Spatial heterogeneity in tumor adhesion qualifies collective cell invasion. Biophysical Journal. 2024 Jun 18;123(12):1635–47. [CrossRef]
- Jain R, Mallya MV, Amoncar S, Palyekar S, Adsul HP, Kumar R, Chawla S. Seroprevalence of SARS-CoV-2 among potential convalescent plasma donors and analysis of their deferral pattern: Experience from tertiary care hospital in western India. Transfusion Clinique et Biologique. 2022 Feb 1;29(1):60–4. [CrossRef]
- Jarrett AM, Lima EABF, Hormuth DA, McKenna MT, Feng X, Ekrut DA, Resende ACM, Brock A, Yankeelov TE. Mathematical Models of Tumor Cell Proliferation: A Review of the Literature. Expert Rev Anticancer Ther. 2018 Dec;18(12):1271–86. [CrossRef]
- Li S, Wang S, Zou X. Data-driven mathematical modeling and quantitative analysis of cell dynamics in the tumor microenvironment. Computers & Mathematics with Applications. 2022 May 1;113:300–14. [CrossRef]
- Padder A, Rahman Shah TU, Afroz A, Mushtaq A, Tomar A. A mathematical model to study the role of dystrophin protein in tumor micro-environment. Sci Rep. 2024 Nov 13;14(1):27801. [CrossRef]
- Mahlbacher G, Curtis LT, Lowengrub J, Frieboes HB. Mathematical modeling of tumor-associated macrophage interactions with the cancer microenvironment. J Immunother Cancer. 2018 Dec 1;6(1):10.
- Szabó A, Merks RMH. Cellular Potts Modeling of Tumor Growth, Tumor Invasion, and Tumor Evolution. Front Oncol [Internet]. 2013 Apr 16 [cited 2025 Apr 6];3. Available from: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2013.00087/full. [CrossRef]
- Zhang Y, Wang K, Du Y, Yang H, Jia G, Huang D, Chen W, Shan Y. Computational Modeling to Determine the Effect of Phenotypic Heterogeneity in Tumors on the Collective Tumor–Immune Interactions. Bull Math Biol. 2023 May 4;85(6):51. [CrossRef]
- Subhadarshini S, Sahoo S, Debnath S, Somarelli JA, Jolly MK. Dynamical modeling of proliferative-invasive plasticity and IFNγ signaling in melanoma reveals mechanisms of PD-L1 expression heterogeneity. J Immunother Cancer. 2023 Sep;11(9):e006766. [CrossRef]
- Zhao B, Pritchard JR, Lauffenburger DA, Hemann MT. Addressing genetic tumor heterogeneity through computationally predictive combination therapy. Cancer Discov. 2014 Feb;4(2):166–74.
- Qian J, Shao X, Bao H, Fang Y, Guo W, Li C, Li A, Hua H, Fan X. Identification and characterization of cell niches in tissue from spatial omics data at single-cell resolution. Nat Commun. 2025 Feb 16;16(1):1693.
- Carranza FG, Diaz FC, Ninova M, Velazquez-Villarreal E. Current state and future prospects of spatial biology in colorectal cancer. Front Oncol. 2024 Dec 3;14:1513821. [CrossRef]
- Fetahu IS, Esser-Skala W, Dnyansagar R, Sindelar S, Rifatbegovic F, Bileck A, Skos L, Bozsaky E, Lazic D, Shaw L, Tötzl M, Tarlungeanu D, Bernkopf M, Rados M, Weninger W, Tomazou EM, Bock C, Gerner C, Ladenstein R, Farlik M, Fortelny N, Taschner-Mandl S. Single-cell transcriptomics and epigenomics unravel the role of monocytes in neuroblastoma bone marrow metastasis. Nat Commun. 2023 Jun 26;14(1):3620. [CrossRef]
- Cilento MA, Sweeney CJ, Butler LM. Spatial transcriptomics in cancer research and potential clinical impact: a narrative review. J Cancer Res Clin Oncol. 2024 Jun 8;150(6):296.
- Ayestaran I, Galhoz A, Spiegel E, Sidders B, Dry JR, Dondelinger F, Bender A, McDermott U, Iorio F, Menden MP. Identification of Intrinsic Drug Resistance and Its Biomarkers in High-Throughput Pharmacogenomic and CRISPR Screens. Patterns (N Y). 2020 Jul 2;1(5):100065.
- Min DJ, Zhao Y, Monks A, Palmisano A, Hose C, Teicher BA, Doroshow JH, Simon RM. Identification of pharmacodynamic biomarkers and common molecular mechanisms of response to genotoxic agents in cancer cell lines. Cancer Chemother Pharmacol. 2019 Oct;84(4):771–80. [CrossRef]
- Al-Harazi O, Kaya IH, El Allali A, Colak D. A Network-Based Methodology to Identify Subnetwork Markers for Diagnosis and Prognosis of Colorectal Cancer. Front Genet. 2021;12:721949. [CrossRef]
- Partin A, Brettin TS, Zhu Y, Narykov O, Clyde A, Overbeek J, Stevens RL. Deep learning methods for drug response prediction in cancer: Predominant and emerging trends. Front Med [Internet]. 2023 Feb 15 [cited 2025 Apr 6];10. Available from: https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1086097/full.
- Ali M, Aittokallio T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys Rev. 2018 Aug 10;11(1):31–9. [CrossRef]
- Rees MG, Brenan L, do Carmo M, Duggan P, Bajrami B, Arciprete M, Boghossian A, Vaimberg E, Ferrara SJ, Lewis TA, Rosenberg D, Sangpo T, Roth JA, Kaushik VK, Piccioni F, Doench JG, Root DE, Johannessen CM. Systematic identification of biomarker-driven drug combinations to overcome resistance. Nat Chem Biol. 2022 Jun;18(6):615–24. [CrossRef]
- Gondal MN, Butt RN, Shah OS, Sultan MU, Mustafa G, Nasir Z, Hussain R, Khawar H, Qazi R, Tariq M, Faisal A, Chaudhary SU. A Personalized Therapeutics Approach Using an In Silico Drosophila Patient Model Reveals Optimal Chemo- and Targeted Therapy Combinations for Colorectal Cancer. Front Oncol. 2021 Jul 16;11:692592. [CrossRef]
- Wu Z, Huang D, Wang J, Zhao Y, Sun W, Shen X. Engineering Heterogeneous Tumor Models for Biomedical Applications. Adv Sci (Weinh). 2023 Nov 9;11(1):2304160. [CrossRef]
- Tripathi S, Augustin AI, Dunlop A, Sukumaran R, Dheer S, Zavalny A, Haslam O, Austin T, Donchez J, Tripathi PK, Kim E. Recent advances and application of generative adversarial networks in drug discovery, development, and targeting. Artificial Intelligence in the Life Sciences. 2022 Dec 1;2:100045. [CrossRef]
- Bacon ER, Ihle K, Guo W, Egelston CA, Simons DL, Wei C, Tumyan L, Schmolze D, Lee PP, Waisman JR. Tumor heterogeneity and clinically invisible micrometastases in metastatic breast cancer—a call for enhanced surveillance strategies. npj Precis Onc. 2024 Mar 29;8(1):1–11. [CrossRef]
- McKigney N, Seligmann J, Twiddy M, Bach S, Mohamed F, Fearnhead N, Brown JM, Harji DP. A qualitative study to understand the challenges of conducting randomised controlled trials of complex interventions in metastatic colorectal cancer. Trials. 2025 Mar 19;26(1):98. [CrossRef]
- Samadian H, Jafari S, Sepand MR, Alaei L, Sadegh Malvajerd S, Jaymand M, Ghobadinezhad F, Jahanshahi F, Hamblin MR, Derakhshankhah H, Izadi Z. 3D bioprinting technology to mimic the tumor microenvironment: tumor-on-a-chip concept. Materials Today Advances. 2021 Dec 1;12:100160. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



