
Submitted:
30 June 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
Alterations in the hypothalamic-pituitary-adrenal (HPA) axis and associated changes in circulating levels of glucocorticoids are integral to an organism's response to stressful stimuli. Glucocorticoids acting via glucocorticoid receptors (GR) play a role in fertility, reproduction, placental function, and fetal development. GRs are ubiquitously expressed throughout the female reproductive system and regulate normal reproductive function. Stress induced glucocorticoids have been shown to inhibit reproduction and affect female gonadal function by supressing the hypothalamic-pituitary-gonadal (HPG) axis at each level. Furthermore, during pregnancy, a mother's exposure to prenatal stress or external glucocorticoids can result in long-lasting alterations to the fetal HPA and neuroendocrine function. Several GR isoforms generated via alternative splicing or translation initiation from the GR gene have been identified in the mammalian ovary and uterus. The GR isoforms identified include the splice variants, GRα and GRβ, GRγ and GR-P. Glucocorticoids can exert both stimulatory and inhibitory effects and both pro- and anti-inflammatory functions in the ovary, in vitro. In the placenta, thirteen GR isoforms have been identified in human, guinea pig, sheep, rat and mouse indicating it is conserved across species and may be important in mediating a differential response to stress. Distinctive responses to glucocorticoids, differential birth outcomes in pregnancy complications, and sex-based variations in the response to stress could all potentially be dependent on a particular GR expression pattern. This review provides an overview of the structure and function of the GR in relation to female fertility and reproduction and discusses the changes in GR and glucocorticoid signalling during pregnancy. This review will delve into the existing understanding of GR isoforms and explore the possible roles that these distinct isoforms may have in regulating glucocorticoid signalling, along with their impact on gonadal activity, placental function, and fetal growth.
Keywords:
glucocorticoid receptor
; reproduction
; pregnancy
; stress
1. Glucocorticoids and hypothalamic-pituitary-adrenal axis
Glucocorticoids, named for their effects on glucose metabolism, play a crucial role in regulating metabolic homeostasis, the cardiovascular system, cell proliferation and survival, growth, cognition and behaviour, immune function, and reproduction [1]. Glucocorticoids are steroid hormones that are synthesized and released by the adrenal cortex under the regulation of the hypothalamic-pituitary-adrenal (HPA) axis (Figure 1) [2]. In the presence of psychological or physiological stress, hypothalamic neurons in the paraventricular nucleus (PVN) synthesize and release corticotropin-releasing hormone (CRH) into the hypophysial portal vessels, which stimulates the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary into the bloodstream [3]. ACTH primarily acts on the adrenal cortex, stimulating the synthesis and secretion of glucocorticoids from the zona fasciculata [2]. Glucocorticoids can inhibit CRH expression and secretion and ACTH output via negative feedback mechanisms [4,5]. Under normal, unstressed conditions, glucocorticoids are released in a circadian and ultradian rhythm. Exposure to acute stressors activates the HPA axis and leads to a temporary increase in glucocorticoids, which returns to baseline following resolution of the stressor; conversely, chronic stress can lead to disruption of the circadian and derangement of ultradian rhythm of glucocorticoid secretion [2,6]. While glucocorticoids are typically adaptive to stress, excessive or inadequate activation of the HPA axis may contribute to the development of pathological conditions [6].
2. Immunological and therapeutical function of glucocorticoids
The immunomodulatory actions of glucocorticoids are highly complex and include both immune stimulatory and immunosuppressive actions. Glucocorticoids are widely prescribed for their anti-inflammatory and immunosuppressive properties to treat inflammatory conditions, autoimmune disorders, haematological malignancies, and many other diverse uses [7]. Physiologically, glucocorticoids enhance innate immunity while limiting inflammation, supporting pathogen clearance and preventing excessive tissue damage, to restore homeostasis following an inflammatory response [8]. These dichotomous impacts on inflammation are dictated by the cellular lineage and activation state, stage of the inflammatory response, and glucocorticoid concentration [9]. In the innate immune response, glucocorticoids upregulate pattern-recognition receptors such as toll-like receptor 2 (TLR2) and NOD-like receptors (NLRP1, NLRP3, NLRC4) [10] to improve detection of pathogen-associated or damage-associated molecular patterns (PAMP/DAMPs). Downstream of these receptors, pro-inflammatory cytokines (such as IL-6, IL-1β and TNF-α) are produced under the control of transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1 (AP-1) [8,11]. These pro-inflammatory cytokines initiate an immune and acute-phase response which promotes glucocorticoid secretion, creating a feedforward mechanism that limits inflammation [11]. Glucocorticoids directly inhibit AP-1 and NF-κB, and upregulate their inhibitors, effectively dampening the inflammatory response and contributing to resolution of inflammation [8]. Glucocorticoids further influence the resolution phase by inhibiting neutrophil recruitment, promoting apoptosis in neutrophils and lymphocytes, and activating macrophages at the injury site for tissue repair [8]. However, when administered in therapeutic doses, glucocorticoids primarily exhibit immunosuppressive effects. These effects include inhibiting leukocyte migration into tissues, inducing eosinophil apoptosis and preventing neutrophil emergence from marrow, reducing circulating lymphocyte counts (more so T cells than B cells) through apoptosis and sequestration, and subsequently reducing immunoglobulin levels [12,13,14,15].
3. Glucocorticoid Receptor
The cellular action of glucocorticoids are mediated by an intracellular protein, the glucocorticoid receptor (GR) [16]. A number of GR isoforms, originating from different splice variants and transcription initiation sites, have been discovered. These isoforms diversify the range of glucocorticoid actions and play a role in determining cellular responsiveness to glucocorticoids [17]. While not the only determinant of glucocorticoid action, the composition of different glucocorticoid receptor isoforms differs between cell types and likely mediates differences in cellular activation state. Furthermore, differences in cellular responses to high or low glucocorticoid doses, natural or synthetic glucocorticoids as well as changes that occur over time, are likely mediated by glucocorticoid receptor subtypes.
The GR is a member of the highly conserved nuclear receptor subfamily 3 [18]. All members of this superfamily share a similar domain structure, consisting of an N-terminal ligand-independent transactivation (AF1) domain, a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) [19]. Apart from containing the ligand-binding pocket, the LBD also contains crucial sequences for receptor dimerization and nuclear localization, as well as a second transactivation domain (AF2) that facilitates interactions with coregulators in a ligand-dependent manner [19]. A small flexible hinge region separates the DBD and LBD, which is involved in conformational changes induced after ligand-binding [19].
The human GR, encoded by the NR3C1 gene, is composed of nine exons [19]. Exon 1 forms the 5′-untranslated region, while exon 2 encodes the N-terminal domain (NTD), which is poorly conserved across nuclear receptors [19]. Exons 3 and 4 constitute the DBD, the most conserved domain in the nuclear receptor family, while exons 5-9 encode the hinge region and ligand-binding domain (LBD) [19]. The NR3C1 gene undergoes alternative splicing, leading to the production of human GRα, GRβ, GRγ, GR-A, and GR-P transcriptional isoforms (Figure 2) [19,20,21]. Most research has focused on the glucocorticoid signalling through GRα and GRβ, proteins identical up to amino acid 727 but differing in their C-terminal exon 9 [22]. The GRβ variant lacks sequences encoding the 11th and 12th helices of the LBD, making it incapable of binding glucocorticoids [22]. Though initially thought to serve primarily as a dominant negative regulator of GRα, transcriptional analysis shows that GRβ has inherent transcriptional activity when overexpressed [23,24,25].
The sequence of GR-γ differs due to an arginine residue included between exons 3 and 4 of the DBD, resulting from the use of an alternative intronic splice donor site [26]. The insertion of a single amino acid significantly reduces the transcriptional activity of GR-γ, demonstrating the functional significance of this region [27,28]. Moreover, changes in the DBD of GR-γ alter its DNA binding sequence specificity, leading to a unique transcriptome [29,30]. The in vivo functions of the GR-A and GR-P isoforms, which contain incomplete LBDs, are not well-understood, but both have been demonstrated in glucocorticoid resistant malignant cell lines [21,31].
All GR splice variants could possibly generate additional isoforms through alternative translation initiation mechanisms, these have only been described for GRα to date. In exon 2, eight highly conserved AUG start codons generate various GR isoforms through ribosomal leaky scanning and shunting, including GRα-A, -B, -C1, -C2, -C3, -D1, -D2, and -D3 (Figure 2). The relative expression of both splice and translation GR isoforms can vary among tissues and within individual cells, influencing their transcriptional potential [32]. Each GR isoform is also subject to several post-translational modifications, such as phosphorylation, acetylation, ubiquitination, and sumoylation, further influencing receptor activity [1].
The traditional explanation for GR signalling focused on glucocorticoids passing through the plasma membrane and binding to GR anchored within the cytoplasm which induced translocation to the nucleus to regulate gene transcription. More recent discoveries have expanded our understanding of GR function to include a multitude of mechanisms of GR impacting gene regulation, the varying impact of GR isoforms on regulating cellular response, and non-genomic effects of GR. There are methodological limitations in identifying and understanding the effects of individual GR isoforms. These limitations include a lack of specific antibodies for each GR isoform, leading to the use of semi-quantitative technique of western blot for the characterization of the GR isoform profile in different cell types and tissues, distinguishing different isoforms by molecular weight. Furthermore, identifying the role of each specific GR isoform has been limited to the use of overexpression experiments in GR null cell lines [32], which does not account for the interaction of multiple GR isoforms that are expressed endogenously.
4. Glucocorticoid Bioavailability
A number of factors can impact glucocorticoid bioavailability to the GR. Roughly 95% of circulating glucocorticoids are bound to cortisol-binding globulin (CBG) or albumin following secretion and the expression of these binding proteins impacts the level of freely available, biologically active glucocorticoid [34]. Endogenous glucocorticoids in circulation have an inconsistent half-life (66–120 minutes), however, those bound to CBG tend to have a longer half-life compared to the unbound hormone [34]. Given the lipophilic nature of glucocorticoids, free hormone can traverse the plasma membrane through simple diffusion. In cells that express multi-drug resistance p-glycoprotein, intracellular glucocorticoid concentration can be limited through their active expulsion [2]. Once inside the cell, glucocorticoids' conversion to active (in humans, cortisone to cortisol) by the enzyme 11β-HSD 1 or to inactive forms (cortisol to cortisone) by 11β-HSD 2 regulates the availability of ligands to the GR [2]. Through these mechanisms, glucocorticoid availability to activate GR, and hence downstream signalling can be finely tuned [2].
5. Glucocorticoid Receptor Signalling
More recent understanding of glucocorticoid function has demonstrated that they act through both genomic (predominant) and non-genomic pathways. The genomic pathway is mediated by GR (Figure 3). In the absence of glucocorticoid, GR exists principally anchored in the cytoplasm as part of a large complex of proteins, including chaperones (hsp90, hsp70, and p23) and immunophilins (FKBP51 and FKBP52) [50], which help maintain GR in a conformation that allows it to bind to ligands with high affinity [35]. Once glucocorticoids bind to GR, receptor conformational change leads to the dissociation of the protein complex and the exposure of 2 nuclear localization signals [33]. The ligand-bound GR is then quickly transported into the nucleus through nuclear pores. Inside the nucleus, the GR binds directly to glucocorticoid response elements (GREs) as a dimer and activates the expression of target genes [33]. This binding leads to further conformational changes in GR, allowing it to recruit coregulators and chromatin-remodelling complexes that influence the activity of RNA polymerase II and activate gene transcription and repression [33]. Binding to negative glucocorticoid-responsive elements (nGREs) can recruit corepressors (NCoR1 and SMRT) and histone deacetylases (HDACs) to repress the activity of target genes [36]. In addition to its direct interaction with GREs, the GR can also interact with members of the signal transducer and activator of transcription (STAT) family to enhance transcription of specific target genes [36]. The anti-inflammatory effects of glucocorticoids are mainly mediated by a negative regulatory mechanism called trans-repression. In this, ligand-bound GR is recruited to genes by directly interacting with DNA-bound transcription factors, particularly NF-κB and activator protein-1 (AP-1) [37]. There are a number of proteins within the cell which can interact with the GR and potentiate trans-repression. For instance, O-linked-N-acetylglucosamine transferase (OGT), a placental stress biomarker, can interact with GR and impact NF-kB mediated transcription [38]. GR directly binds to the p65 subunit of NF-kB and the Jun subunit of AP-1, inhibiting their transcriptional activation [37]. For some genes, GR functions in a composite manner, binding directly to a GRE and physically associating with AP1 or NF-kB bound to a neighbouring site on the DNA, while for others, GR binds to transcription factors without interacting directly with DNA [33].
The non-genomic effects of glucocorticoids, occur without requiring gene transcription, are rapid and mediated through physiochemical interactions of cytosolic or membrane-bound GR [39]. These effects happen within seconds to minutes of activation and involve the activity of kinases such as phosphoinositide 3-kinase, AKT, and mitogen-activated protein kinases (MAPKs) [40]. Additionally, when glucocorticoids bind to GR, accessory proteins are released, which participate in secondary signalling pathways [40]. This non-genomic signalling of the glucocorticoid receptor increases the complexity and diversity of biological actions that are dependent on glucocorticoids [33].
6. Glucocorticoids on hypothalamic-pituitary-gonadal axis
The hypothalamus and pituitary are also central to reproductive physiology, serving as a hub for both receiving and delivering endocrine signals [41]. The hypothalamus generates and releases gonadotropin-releasing hormone (GnRH), which travels through the hypophyseal portal system to reach the anterior pituitary [42]. There, GnRH stimulates the synthesis and release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the gonadotrophs, leading to the secretion of estradiol and progesterone from the ovaries [43]. A regulatory negative feedback mechanism exists in which estradiol from the ovaries inhibits the secretion of GnRH and thus the production of FSH and LH [43]. Increased glucocorticoid exposure, whether resulting from stress or exogenous administration, can lead to significant reproductive disturbances through their notable impacts on the hypothalamus and pituitary [44,45,46]. Glucocorticoids influence the HPG axis by inhibiting the release of GnRH from the hypothalamus and the synthesis and release of gonadotropins from the pituitary [44,46].
The presence of GR in the hypothalamus implies that glucocorticoids could have direct effects [47]. Indeed, dexamethasone-induced GR signalling in hypothalamic cell lines that secrete GnRH suppresses GnRH promoter activity and subsequent GnRH mRNA levels in vitro by interacting with a multiprotein complex at negative GRE sites [48]. A study analysing the reproductive function in adult male rats treated with corticosterone over an extended period reported decreased hypothalamic GnRH mRNA and decreased serum LH but not FSH secretion [49]. The same study examining the acute effects of dexamethasone on reproductive function in adult female rats reported a significant decrease in pituitary FSHβ but not LHβ mRNA levels [49]. The findings from this study suggest that chronic increases in levels of glucocorticoid/stress suppress gonadotropin levels by reducing the expression of GnRH levels while acute glucocorticoid administration prevents the gonadotropin secretion via other processes, including the reduction of FSHβ mRNA levels. However, this effect of acute stress and short-term increases in glucocorticoid level on gonadotrophin secretion can vary, either inhibiting or stimulating it. GnRH stimulates the production and release of LH and FSH by binding to the GnRHR in pituitary gonadotropes [50,51]. Evidence from the mouse pituitary gonadotrope cell line LβT2 treated with dexamethasone suggests that glucocorticoids could directly induce the expression of the GnRHR gene, leading to increases in LH and FSH secretion [52]. In addition, in primary cultured cells from the anterior pituitary, glucocorticoids have been found to increase FSHβ mRNA levels [49,53]. These are inconsistent with the findings from Gore et al [49] which may suggest that the way glucocorticoids control the expression of gonadotropin synthesis is dependent on the context. Our understanding of the regulation of gonadotrophins by glucocorticoids is presently based on findings from cell line studies, which may not accurately depict the regulation by endogenous glucocorticoid in vivo [46].
Besides directly controlling GnRH, GnRHR, and pituitary gonadotropes, glucocorticoids can also regulate their inhibitory effects on HPG function via novel intermediaries. Glucocorticoids can inhibit GnRH expression by regulating the expression of two neuropeptides, kisspeptin (KISS1) and gonadotropin-inhibitory hormone (GnIH) [46]. These neuropeptides exert contrasting impacts on GnRH secretion from the hypothalamus and react to elevated glucocorticoid levels. KISS1 promotes the GnRH secretion via its receptor KISS-1R, co-expressed in GnRH neurons. KISS1 neurons located in the anteroventral periventricular nucleus and the periventricular nucleus continuum of the preoptic area in the hypothalamus express GR, indicating that glucocorticoids can directly act on these neurons [54]. Corticosterone administration in mice inhibited ovarian cyclicity by disrupting the preovulatory hormonal cascade required for the LH surge [55]. The study also reported a significant decrease in the activation and percentage of KISS1 neurons as well as the expression of KISS1 mRNA in the hypothalamus during an estradiol-induced LH surge [55]. This suggests that glucocorticoids can suppress the HPG axis by inhibiting the expression of KISS1 neurons. GnIH neurons suppress the functions of GnRH and KISS1 neurons, and gonadotrophs [56]. Both acute and chronic stress in sheep lead to an increase in expression and activity of GnIH neurons and increased the interaction between GnIH fibres and GnRH cells [57]. However, stress had no effect on GnIH levels in the hypophysial portal blood [57]. Moreover, this effect on the GnIH gene expression disappeared after adrenalectomy, implying that adrenal-derived factors, most likely corticosterone mediated the stress-induced inhibition of reproductive functions [57]. GnIH neurons co-express GR, and the exposure of quail and rat hypothalamic neuronal cells to corticosterone resulted in increased GnIH expression [57]. This increase was facilitated through glucocorticoid response elements (GREs) in the promoter region of GnIH [57]. In short, glucocorticoids can indirectly influence the reproductive functions of the HPG axis by regulating GnRH expression and secretion by KISS1 and GnIH.
7. Effect of glucocorticoids on reproductive organs
As glucocorticoids are known to be a key regulator of basal and stress-related homeostasis, it is highly likely that stress-related increases in glucocorticoid levels could contribute to decreased fertility [44]. Stress-related compromises to reproductive function can be reported across various species. Mammals such as rats [58,59,60], along with birds, reptiles [61,62,63,64] all exhibit a reduction in both male and female reproductive abilities in response to physical stress [3]. The response to stress in humans is also detrimental to reproductive function throughout the life cycle. For example, high perceived stress during pregnancy is a risk factor for preterm labour and poor outcomes in offspring [65,66]. For female athletes, there is a high prevalence of delayed puberty, menstrual disorders, and long-term secondary amenorrhoea, especially those participating in aesthetic sports and endurance sports [67,68]. This reproductive dysfunction could be caused by the alteration of HPG axis activity due to excess cortisol associated with physiological and psychological stress on the body. Glucocorticoids are also known to interfere with sexual maturation as patients with hypocortisolism (Addison's disease) and hypercortisolism (Cushing's syndrome) have altered onset of puberty [69,70]. A study revealed a positive correlation between circulating glucocorticoid levels and age of puberty onset in prepubertal girls [70]. This is consistent with findings from animal studies [71,72,73]. Studies in mice and rats report delayed onset of puberty in offspring of mothers subjected to stress or with synthetic glucocorticoid administration [73,74,75]. Prepubertal exposure to synthetic glucocorticoids delayed onset of vaginal opening, an indicator of puberty in female rats [76].
7.1. Ovary
Apart from influencing the ovarian cycle via the hypothalamus and pituitary, glucocorticoids can also directly affect ovarian functions by controlling the activities of granulosa cells, oocytes, cumulus cells, and luteal cells through GR [55]. It was previously thought that the ovary does not produce glucocorticoids locally. Hence, it was presumed that glucocorticoids in the ovary were transported from the adrenal glands via the bloodstream [77]. These glucocorticoids, probably inactive (cortisone), were then proposed to diffuse into the granulosa cells of the periovulatory follicle, where 11β-HSD 1 converted it to cortisol which could then exert its effects via co-localised GR [77]. However, a recent study by Jeon et al [78] demonstrated human primary luteinizing granulosa cells produce cortisol in vitro. The mRNA levels of CYP11B1 and CYP21A2, two enzymes required for the biosynthesis of cortisol, were detected in primary cultured human granulosa/lutein cells (hGLC) and their levels were found to increase following ovulatory administration of human chorionic gonadotropin (hCG; as a substitute for LH) [78]. CYP21A2, an enzyme necessary for the metabolism of 17α-hydroxyprogesterone into 11-deoxycortisol, and CYP11B1, an enzyme responsible for the conversion of 11-deoxycortisol into cortisol, are both required for the synthesis of cortisol from progesterone (Figure 4) [78]. This is consistent with findings from Amin et al [79] which reported the levels of two cortisol precursors, 11-deoxycorticosterone and 11-deoxycortisol, in the follicular fluid to be positively correlated with the lipid content in human luteinized granulosa cells (LGCs) and demonstrated the expression of CYP21A2 mRNA in LGCs in vitro. Given that during ovulation, human granulosa cells generate a substantial amount of progesterone, the presence of enzymes that convert progesterone to cortisol suggest that these cells could produce cortisol [78]. However, it is still likely that the majority of the cortisol acting on the ovaries is derived from the adrenal glands instead of being locally produced as this evidence is based on in vitro studies and might not accurately represent in vivo physiology.
Glucocorticoid concentrations are intracellularly regulated in the ovary by 11β -HSD 1 and 2. 11β-HSD has been shown to be present in various ovarian cell types such as oocytes, cumulus cells, granulosa cells, theca cells, granulosa-lutein cells, corpus luteum, and ovarian surface epithelium [80,81,82]. During the LH surge, there is a rapid increase in the production of progesterone and cortisol. Before the LH surge, follicular levels of inactive glucocorticoids (cortisone) are higher than those of cortisol [83]. However, post-surge this reverses, with the concentration of cortisol being 4.5-fold higher than that of cortisone [83]. This switch is proposed to be caused by a change in predominance of 11β-HSD isotype from 2 to 1 [83,84]. In non-luteinized granulosa cells from IVF patients, 11β-HSD 2 was readily detected, but it was not detectable in the luteinizing granulosa cells [85]. More recent studies have also demonstrated a swift decrease in 11β-HSD 2 (mRNA) and increased 11β-HSD 1 (mRNA and protein) levels in human granulosa cells collected after hCG stimulation in comparison to cells obtained prior to the in vivo LH surge [86], as well as in vitro [78]. The increased expression of 11β-HSD 1 persisted in post-ovulatory follicles, suggesting it has a role during ovulation and the initial phase of luteal formation by modulating intracellular glucocorticoid concentrations. Furthermore, 11β-HSD 1 expression was mainly observed in granulosa cells from late ovulatory follicles and granulosa-lutein cells of post-ovulatory follicles, indicating the location of cortisol synthesis [78]. These findings suggests that active glucocorticoid concentrations are reduced during follicular growth and maturation, but they rise during the ovulation process initiated by the LH surge.
Preovulatory follicles undergo an acute inflammatory event during ovulation which is associated with rupture, the subsequent healing process, and metabolic alterations linked with luteinization. Thus, the coinciding increase in expression of anti-inflammatory glucocorticoids [87] by 11β-HSD 1 activation of cortisone to cortisol may be a physiological compensatory mechanism to reduce the ovarian inflammatory process [78]. To determine the effect of cortisol on periovulatory follicles, Jeon et al [78] looked at the effect of ovulatory hCG stimulation on the expression of genes involved in cortisol synthesis. The expression of 11β-HSD 1, NR3C1, and FKBP5 were increased after hCG administration in periovulatory follicles, in vitro and in vivo [78]. Moreover, it was reported through immunohistochemistry that GR was localised in endothelial cells and leukocytes within and near the late and post-ovulatory follicles [78]. These findings suggest that glucocorticoids probably operate during ovulation and the initial stages of corpus luteum formation by exerting their effect directly on follicular cells, endothelial cells, and leukocytes, all of which express GR. Since cortisol plays a role in ovulation and corpus luteum (CL) formation, this could contribute to women with dysregulated levels of cortisol frequently experience menstrual irregularities and anovulatory infertility.
Furthermore, stress-induced glucocorticoid levels have been linked to a decrease in oocyte competence. Chronic heat stress in pigs has been shown to modify ovarian expression of genes involved in steroidogenesis and increase signalling via the insulin-mediated phosphoinositide 3-Kinase (PI3K) pathway [88]. Heat stress can also alter the composition of follicular fluid, thereby changing the oocyte’s microenvironment [89,90]. In mice, the damage to oocyte competence was found to be dependent on the severity of the restraint stress, with more significant effects observed after prolonged stress [91]. Administration of exogenous glucocorticoids in female rats led to a decline in growth factor levels and alterations in the estrogen to progesterone ratio [92].
7.1.1. Dual role of glucocorticoids in the ovary
Despite the studies aimed at determining the role of glucocorticoids in the ovary, their function is not completely understood, with contradictory findings from multiple studies. Several studies have looked at the effect of glucocorticoid administration on mammalian oocytes, and its influence on oocyte maturation, with inconsistent results. In some in vitro mice studies, no effect was found on oocyte maturation when they were subjected to cortisol (up to 28 μM) and dexamethasone (up to 100 μM) [93,94,95,96]. However, in other mice studies, slightly higher concentrations of cortisol (138 μM) and dexamethasone (204 μM) resulted in inhibition of oocyte maturation in vitro [93,95,96]. Similar to studies in mice, cortisol and dexamethasone administration (between 0.26 and 138 μM) inhibited oocyte maturation in porcine oocytes in vitro [97]. Conversely, a study on horses reported similar doses of cortisol (0.28–2.8 μM) to have no effect on oocyte maturation in vitro [98]. Glucocorticoids can either selectively induce or inhibit ovarian steroidogenesis. Dexamethasone administration in rats is associated with altered production of progesterone from granulosa cells by either increasing or decreasing the expression of steroidogenic proteins like steroidogenic acute regulatory protein (StAR) [99,100,101]. This indicates that in vitro glucocorticoids exhibit both stimulatory and inhibitory effects in the ovary. Such discrepancies could be attributed to variations in the administered dosage, the phase of follicular development, or differential expression of GR isoforms.
Similarly, glucocorticoids can have both pro- and anti-inflammatory roles in the ovary [102]. Cortisol treatment has also been shown to decrease germ cell density by increasing the apoptosis of oogonia in developing human ovary, impair oocyte development potential and increase apoptosis of granulosa cells in animal models [103]. However, glucocorticoids have also been reported to inhibit apoptosis triggered by cAMP, p53, and TNF-α stimulation and be cytoprotective in immortalized rat granulosa cells [104]. In primary bovine luteal cells, cortisol has been reported to prevent apoptosis induced by TNF and interferon γ (IFN- γ) by inhibiting the expression of caspase 8 and 3, thus preserving the function of the corpus luteum [105]. The anti-apoptotic effects of glucocorticoids could also contribute to reducing damage associated with follicle rupture. Glucocorticoids, via GR signalling, reduce the expression and function of extracellular matrix metalloproteinase 9 in human ovarian surface epithelial cells which helps inhibit proteolytic injury to the ovarian surface during ovulation with the associated inflammation [106]. These opposing effects of glucocorticoids on the ovary cannot be explained by the expression of a single isoform of GR and could potentially be a result of differential expression of the GR isoforms.
7.1.2. Glucocorticoid receptor isoforms in the ovary
GR is found in the follicles, corpus luteum, and cells of the ovarian surface epithelium in both rats and humans [107]. In human studies, GR protein has been identified in ovarian surface epithelium cells, in vitro [108]. In vivo studies have also found GR protein to be localised in granulosa cells, theca cells, endothelial cells, and leukocytes in blood vessels in the vicinity of late ovulatory and post-ovulatory follicles [78]. The expression levels of GR in the follicle and corpus luteum remains unchanged during the stages of follicular maturation and ovulation in rats [109], implying that the main regulatory processes in these cell types could be determine by the concentration of active glucocorticoids. Current evidence proposes that glucocorticoids have a direct influence on the preliminary phases of reproduction, such as the maturation of oocytes, fertilization, and preimplantation embryo growth [110,111]. However, the observed effects can vary based on factors like the type of glucocorticoid molecule, dosage, the stage of development, and the species under study [111]. As variation in the expression of GR isoforms may lead to distinct cellular responses to glucocorticoids, investigating the expression of various splice and translational isoforms of the GR in oocytes carries significant importance.
Very few studies have looked at the expression of the different isoforms of the GR in mammalian ovary. The ovary is a complex organ with its function highly linked with the active follicles that undergo constant change across the ovarian cycle. This could possibly lead to a constant shift in the expression profiles of the different GR isoforms. A recent study examining mice oocytes found the two splice variants, GRα and GRγ (mRNA) to be highly and consistently expressed, while GRβ and GR-P (mRNA) were undetectable [111]. This study also reported the presence of a doublet band around 94 and 91 kDa in oocytes, and this doublet likely represents mouse isoforms GRα-A and B (GR protein) [111]. Another study [112] examining at the presence of GRβ in various tissues reported the absence of GRβ in SK-OV-3 (ovarian adenocarcinoma) cells in vitro, consistent with the results from the study by Cikos et al [111]. Since GRβ is known to antagonise the effects of GRα, the pro-inflammatory or contradictory effects of cortisol have often been attributed to an increased GRβ:GRα ratio. However, since GRβ is undetectable in the ovary, the varying responses of ovarian cells to high concentrations of glucocorticoids could be attributed to the presence of the other splice or translational isoforms present in the ovary. The detection of various GR isoforms in the ovarian cells needs to be further examined to better elucidate their contribution to ovarian glucocorticoid function.
7.2. Uterus
The influence of glucocorticoids on uterine biology has been typically depicted as antagonism of estrogen activity, but it has been recently studied as an independent moderator of uterine functionality. Dexamethasone impedes estrogen-induced uterine growth and proliferation in mice, and when administered before estradiol-facilitated implantation, can lead to a decrease in implantation sites [113,114]. Gene identification and ontology studies looking at the expression of genes regulated by dexamethasone and estradiol in a human uterine epithelial cell line points to glucocorticoids regulating important biological processes including immune function, cell cycle and embryonic development [115]. For instance, dexamethasone (but not estradiol) upregulated the expression of nuclear factor of kappa light chain inhibitor alpha (NFκB1A), an inhibitor of the NFκB complex. NFκB signalling in the uterus controls the apoptotic function of caspase 3 during pregnancy [115]. The expression of NFκB1A escalates during the late secretory phase, coinciding with increased incidence of apoptosis in the uterus [116,117]. Thus, the regulatory role of glucocorticoid on NFκB1A could contribute to the balance between cell death and survival in the uterus [115]. Furthermore, expression of the gene left-right determination factor 1 (LEFTY1) was upregulated with dexamethasone treatment (restored with estradiol administration) [115]. LEFTY1 is a gene that encodes a soluble cytokine of the TGF-β superfamily [118]. When LEFTY1 is introduced into the uterine horn of pregnant mice, it significantly reduces uterine decidualization and results in a decreased litter size [119]. Thus, glucocorticoid regulation of key fertility genes, like LEFTY1, offers additional evidence that glucocorticoids may function as a bridge between the immune and reproductive systems in the uterus [115].
Dexamethasone administration in mice also singularly regulates the expression of genes associated with cellular development, proliferation, and signalling. In the uterus of neonatal mice, dexamethasone reduces epithelial cell proliferation through mechanisms that are both dependent on and independent of GR [120]. Human endometrial cells that have undergone decidualization also exhibit a distinct transcriptome dependent on the GR [121]. This GR-dependent transcriptome is enriched with Krüppel-associated box domain containing zinc-finger proteins which are a family of transcriptional repressors involved in heterochromatin formation [121]. In fact, decidualization of human endometrial stromal cells was associated with an increase in GR expression [120]. Gene expression analysis of GR knockout HESCs that were undergoing differentiation reported an increase in trimethylated H3K9 levels in decidualizing cells [120]. This suggests that GR can directly manage gene expression and globally modify transcription through the regulation of histone alterations in human uterine cells [120].
The semi-allogenic fetus's immunological tolerance and a successful pregnancy require pre-emptive immune system adjustments that support endometrial receptivity [46]. These changes begin before the implantation of the embryo and escalate throughout the early stages of pregnancy [122,123,124]. During this time, immune cells are mobilized and activated in the uterus, establishing a dynamic immune response that is crucial for implantation and tolerance [46]. When this immune balance is disrupted, it can contribute to complications such as miscarriage, preeclampsia, or premature labour. Glucocorticoids can regulate the uterine immune system by directly or indirectly acting on these immune cells. Studies involving conditional ablation of GR in mouse uteri have suggested the essential role of endometrial glucocorticoid signalling in recruiting appropriate immune cells [125]. In addition, in these GR knockouts, the expression of genes required for implantation was altered. This implies that the glucocorticoid signalling in the uterus might play a critical role in establishing the molecular groundwork that determines uterine receptivity [125].
Administration of exogenous glucocorticoids affects the number and function of uterine immune cells [126]. In the initial stages of pregnancy, uterine natural killer (uNK) cells control trophoblast invasion and changes in the vascular functions that help placental growth [127,128]. GR is expressed in uNK cells, and their cytotoxic activity is responsive to cortisol treatment [129]. The administration of prednisolone in women experiencing recurrent miscarriage correlates with decreased uNK cell counts, however, there is yet to be a demonstrated connection between this glucocorticoid-induced reduction in uNK cells and the success of the pregnancy [130]. Macrophages exist in two distinct forms: an M1, or pro-inflammatory phenotype, and an M2 phenotype associated with tissue remodelling, immunosuppression, and angiogenesis [131]. During the peri-implantation period, endometrial macrophages lean towards the M1 phenotype and shift towards the M2 phenotype post-implantation. [132,133]. GR is expressed in human endometrial macrophages during the secretory phase, which coincides with the uterine receptivity period, implantation, and the menstrual phase, as well as at full term, but the function of glucocorticoid signalling in these macrophages remains largely unexplored [134,135]. Dendritic cells and regulatory T cells play a vital role in determining whether the blastocyst is accepted or rejected. However, the physiological impacts of glucocorticoids on these cell types within the endometrium remain under-researched [136].
In the peripheral immune system, glucocorticoids exhibit a broad range of direct influences on the development and function of immune cells, which can potentially affect the composition of immune cells available for recruitment to the reproductive tract [8]. They restrict the formation of immature dendritic cells from monocytes and inhibit the maturation of immature dendritic cells in vitro [137]. Elevations in serum glucocorticoid levels have been linked to the rapid increase of mature NK cells in peripheral circulation in vivo [138]. Furthermore, glucocorticoids stimulate substantial apoptosis in T and B cells, mature dendritic cells, basophils, and eosinophils [9]. Glucocorticoids suppress the transcription of pro-inflammatory cytokines and chemokines in macrophages and guide macrophages towards the M2 phenotype [126]. Dexamethasone can also trigger components of the inflammasome, thereby enhancing the innate immune response in macrophages [139]. The response of the immune system to glucocorticoids relies on both the physiological or stress-induced hormone levels and the duration of the stimulus. This suggests that the immune response that oversees reproduction is partly managed through physiological glucocorticoid signalling by the GR and is susceptible to hormone fluctuations.
7.2.1. GR isoforms in the uterus
GR is found in the uterus, predominantly within the stromal fibroblasts, lymphocytes, and endothelial cells, with only minimal presence in the glandular epithelium. Contrary to the cyclical changes reported with the estrogen receptor (ER) and progesterone receptor (PR), GR expression remains steady throughout the menstrual cycle [140]. During labour, there is a significant increase in total GR and GRα protein levels, while the levels of GRβ do not undergo any change [141]. Given the limited research on the expression of GR isoforms in the uterus, further studies are necessary to investigate if differential expression of these isoforms is behind the contradictory effects of glucocorticoids reported in the uterus.
8. Pregnancy and Parturition
During the second and third trimesters of pregnancy, the placenta begins to produce CRH, which has the same biological activity as the CRH produced by the hypothalamus (Figure 5) [66]. The placental CRH (pCRH) is secreted into both the maternal and fetal compartments. Unlike its inhibitory effect on the CRH-producing cells of the hypothalamus, cortisol stimulates the synthesis and release of pCRH [66], leading to a shift from negative feed-back regulation of the maternal HPA axis to a positive feed-forward cycle. Consequently, the circulating CRH levels in pregnant women are 1,000 to 10,000 times higher than non-pregnant individuals [142]. Throughout gestation, most of the CRH is inactively bound to the CRH-binding protein (CRH-BP), however, during the third trimester, a spike in CRH occurs without a corresponding increase in CRH-BP levels in the maternal circulation, leading to an increase in the levels of active CRH [143]. It has been proposed that this rise in CRH levels may determine the length of gestation and initiate uterine contractility at the start of labour, acting as a "placental clock" [142]. However, if certain abnormalities occur during pregnancy, such as preeclampsia or maternal stress, these alterations may occur prematurely resulting from excess cortisol, which may lead to adverse maternal and/or fetal outcomes including preterm birth and fetal growth restriction [66].
During pregnancy and parturition, uterine contractility can be separated into at least four stages. The first stage, known as phase 0, is the pregnancy period where the uterus stays relatively quiescent through the action of inhibitors of uterine contractility such as progesterone [144]. In the first phase of parturition [phase 1], the uterus responds to mechanical stretch or uterotrophic priming by activating a set of genes essential for myometrial contraction [144]. As parturition enters phase 2, the uterus is stimulated by various uterotonins such as prostaglandins (PGs), oxytocin, and CRH [144]. The third and final phase involves uterine involution following the delivery of the fetus and placenta, a process primarily influenced by oxytocin [144]. Glucocorticoids help with two major events that lead to parturition [145]. Firstly, glucocorticoids stimulate the production of the placental enzyme cytochrome P450 17α hydroxylase (P450c17), which converts C21 steroids (pregnenolone and progesterone) into C19 steroid aromatase precursors [146]. This triggers a decrease in progesterone alongside an increase in estrogen.
The rising estrogen then primes the myometrium from a state of quiescence to a contractile state by upregulating the expression of contraction-associated proteins (CAPs) such as connexin 43 [Cx43], oxytocin receptor (OTR), and prostaglandin (PG) receptor. After myometrium priming, sufficient uterotonins, such as prostaglandins, are synthesized to complete the feed-forward process of parturition [146]. Apart from initiating myometrial contractions, prostaglandins E2 and F2α (PGE2 and PGF2α) also facilitate cervical ripening, membrane rupture prior to parturition, and promote the expression of placental P450c17 [146]. In humans, prostaglandin production is compartmentalized within the tissues of the pregnant uterus [147]. While chorion and decidua also contribute, amnion is predominantly responsible for the formation of PGE2, and its output surges at labour onset [147]. Glucocorticoids have been found to elevate PGE2 and PGF2α synthesis in the amnion and chorion through inducing the expression of prostaglandin H synthase (PGHS-2), an enzyme involved in the rate-limiting step in prostaglandin synthesis [148,149]. Furthermore, PGE2 and PGF2α enhance 11β-HSD-1 and decrease 11β-HSD-2 activity in chorion, leading to increased cortisol production [150]. These feedback loops serve to increase both local cortisol and local PG concentrations [150]. 15-hydroxyprostaglandin dehydrogenase (PGDH), an enzyme present in the chorion, metabolises PGs generated from the amnion and the chorion and prevents its passage to underlying tissues [147]. Its activity is usually decreased in preterm births and as a result of the reduced metabolic barrier, the PGs are thought to prematurely stimulate myometrial contractility. Glucocorticoids have been shown to decrease the expression and activity of human chorionic PGDH [147].
Apart from stimulating the synthesis and inhibiting the metabolism of PG, glucocorticoids can also promote the rupture of the amnion. Apoptosis of the amnion epithelial layer occurs before the rupture of the fetal membrane, a key factor for both term and preterm delivery. Studies on human amnion epithelial cells in ex vivo cultures have demonstrated that cortisol triggers this apoptosis through the extrinsic apoptotic pathway [151]. Cortisol also reduces the quantity of collagen proteins and lysyl oxidase, an enzyme responsible for collagen cross-linking, in amnion cells through a process called lysosome-mediated autophagy [152]. The tensile strength of the amnion is determined by collagen content and a reduction in collagen content has been observed in women experiencing preterm membrane rupture [153]. Therefore, glucocorticoids' degenerative impact on amnion cells could be a contributing factor to the premature rupture of fetal membranes, leading to preterm labour.
Placental CRH also directly influences the uterus and cervix by increasing the changes instigated by estrogen within these structures. It stimulates dehydroepiandrosterone (DHEA) production from the fetal adrenal gland which is required for the rise in placental estrogen needed to allow for the increase in uterine oxytocin and prostaglandin receptors which allow onset of parturition [154,155]. At the same time cortisol upregulates prostaglandin production from the placenta and fetal membranes to induce membrane rupture and enhance contraction [155].
Numerous studies have been conducted looking at the impact of externally administered glucocorticoids on inducing parturition. In humans, intra-amniotic glucocorticoids can induce labour, whether at >41 weeks gestation with 20 mg of betamethasone [156], or in patients who are >12 days past their expected date of delivery who were given 500 mg of hydrocortisone [157]. In pregnancies involving triplets or quadruplets, maternal administration of betamethasone led to an increase in uterine contractions, premature labour, and alterations in the cervix [158]. Furthermore, the average gestation period in a cohort of term-born infants was reduced by an average of 4 days in pregnant women who received glucocorticoid treatment (a single dose of 12 mg of dexamethasone or betamethasone given intramuscularly twice over a 24-hour period) during their 30th week of pregnancy as opposed to those who didn't undergo glucocorticoid treatment [159]. Another concern relates to the excessive administration of synthetic glucocorticoids to stimulate fetal lung maturation in women at risk of preterm labour [160]. Despite numerous beneficial effects of endogenous glucocorticoids, such fetal organ maturation [160], exogenously administered corticosteroids given to pregnant women at risk of preterm labour [158] and to animals [161] have been demonstrated to stimulate uterine activity. Thus, it is clear that both term and preterm births are a result of processes leading to an increase in PG production, with glucocorticoids playing an important part.
8.1. Effect of excess cortisol on myometrial contractility during parturition
Increased levels of cortisol due to maternal stress, or an overactivated fetal HPA axis could lead to increased myometrial contractility and increased chance of preterm birth.
Contrary to expectations, stress endured either prior to pregnancy or during gestation does not predict a preterm birth outcome, despite numerous studies suggesting an association. Women experiencing preterm labour often show significantly elevated CRH levels in comparison to controls at the same gestational stage [154,162]. Interestingly, these heightened CRH levels can emerge weeks ahead of preterm labour onset [144]. Under normal circumstances, cortisol levels increase towards the final stages of gestation and during the initiation of labour [102,154,162]. However maternal stress and high cortisol can lead to activation of labour-related mechanisms in the placental and fetal membranes, potentially triggering labour prematurely [154,162]. Understanding how stress can induce preterm labour in some women and not others may be related to the expression of different GR isoforms. The differential expression of GR isoforms between individuals may contribute to the risk of preterm delivery under conditions of stress but this needs to be investigated further.
9. Fetus
During pregnancy, the fetus relies heavily on glucocorticoids for appropriate development. The concentrations of cortisol required by the fetus changes as pregnancy advances with the placenta allowing increasing levels of glucocorticoid transfer as the fetus approaches term. Indeed, maternal glucocorticoids contribute to the growth, development, and survival of the fetus in all mammals [163]. Of particular clinical importance is the role of glucocorticoids in ensuring appropriate lung maturation, but they are also vitally important for development of the kidney, heart, adrenal, genital systems and almost all other fetal tissues [164]. However, exposure to high levels of cortisol due to maternal stress can have significant consequences on fetal development [163]. While some of the negative effects of elevated maternal glucocorticoids are caused by disruption to placental formation [165,166,167], they can also directly impact formation of key systems including the cardiovascular system, kidney, brain, and immune system [168,169,170]. Consequently, it can elevate the risk of future health issues such as cardiovascular, kidney, neurological, respiratory, and metabolic disorders, along with the likelihood of allergies, in the child's later life [171]. Synthetic glucocorticoids are also commonly administered in women who are at risk of giving birth prematurely, as well as those with certain medical conditions like asthma, systemic lupus erythematosus and hyperemesis gravidarum [172]. Excessive exposure to synthetic glucocorticoids during gestation can have far-reaching impacts on an individual's health throughout their lifespan. While previous reviews have focused on describing the wide-ranging programmed effects of glucocorticoids [172], below are a few examples. Elevations in maternal glucocorticoids have been shown to program an altered stress response [173], motor developmental delays, and higher blood pressure [174].
Studies on rodents have also shown that administering synthetic glucocorticoids during pregnancy can lead to sex-specific changes in placental development [166,175] and result in negative health outcomes for offspring [176,177]. For example, dexamethasone administration in mice led to reduced placental size and reduced fetal growth in female placentas [176]. Similarly, in sheep studies, exogenous administration of betamethasone led to reduced fetal growth [176], increased placental apoptosis [178], and decreased levels of insulin-like growth factors [178], suggesting that excess maternal glucocorticoids can impact fetal growth and nutrient transport through changes in the placenta.
9.1. Sex differences
There is a large body of evidence indicating that the effects of prenatal stress on offspring differs between males and females [179]. For example, in asthmatic mothers, increased cortisol levels and decreased cortisol metabolism by 11β-HSD2 in the placenta were associated with reduced birth weights and suppressed fetal adrenal function only in female fetuses [180]. Animal studies have also shown that maternal stress or exposure to cortisol can cause sex-specific changes in placental dysfunction [165,181] and negative health outcomes for offspring [181,182]. The female placenta appears to adapt to fluctuations in glucocorticoid levels, as indicated by alterations in cortisol metabolism, placental cytokine expression, IGF axis signalling, adrenal functionality, and growth [183,184]. In contrast, the male placenta seems less responsive to changes in glucocorticoid levels, with no changes in cortisol regulated pathways [182]. This lack of adaptation in the male placenta is associated with a higher likelihood of intrauterine growth restriction, preterm birth, or stillbirth [184]. On the other hand, female placentas adapt in a way that may lead to reduced growth but enhanced survival [182].
The female placenta has been observed to potentially allow greater glucocorticoid exposure due to the altered activity of enzymes responsible for cortisol metabolism, including 11β-HSD1 2 [183,185]. Intriguingly, unique transcriptional profiles emerge from the differential expression of GR isoforms in response to maternal dexamethasone exposure, and this expression varies between male and female mouse placentas [32,186].
Research also suggests that the impacts of prenatal maternal stress are not only sex-specific but can also extend beyond the neonatal period [187]. For instance, exposure to stress during fetal life in females has lingering effects into early childhood and adolescence, as evidenced by increased anxiety levels, compromised executive function, and neurologic markers linked to these behaviours [188]. Early exposure to high maternal cortisol levels in pregnancy led to significantly larger amygdala and increased anxiety levels in female offspring, but not in males [188]. Additionally, maternal stress correlated with negative emotionality in female offspring, but this association was not found in males [189]. This evidence suggests that exposure to stress in early life can lead to sex-specific outcomes in fetal programming. Such programmed disease outcomes have even been shown to have impacts that persist in the next generation [190,191]. Thus, to understand how excess cortisol or synthetic glucocorticoids affect fetal development and mediate sex specific changes in fetal outcome and programming, it is important to study the glucocorticoid receptor and its downstream signalling pathways [179,192].
9.2. Glucocorticoid receptor isoform in the placenta
There are thirteen glucocorticoid receptor (GR) protein isoforms in the placenta of the human [187,188], guinea pig [189], sheep [190], mouse [107] and the rat [191] that vary in relation to gestational age at delivery, fetal sex, circulating glucocorticoid concentration, fetal growth and maternal stress. Changes in GR isoforms in the placenta may be a novel mechanism to explain how different placentae have varied responses to pregnancy complications including pregnancies complicated with asthma. For instance, an increase in cortisol levels is associated with a rise in the expression of GRβ protein in the cytoplasm of male placentae from pregnancies complicated by asthma and in both the cytoplasm and nucleus of placentae in small for gestational age (SGA) pregnancies [187]. Thus, in scenarios of increased cortisol exposure, males may establish a state of glucocorticoid resistance through the increased expression and nuclear localization of GRβ, which then suppresses the activity of GRα in a dominant-negative manner [187]. This resistance state might also be influenced by the nuclear co-localization of GR-A and GR-P in the male placental trophoblast [187].
The interaction between different GR isoforms could also lead to activation of different downstream pathways and target genes. For example, under basal conditions, GRα and GRβ are co-expressed in neutrophils [192]. However, when these cells are exposed to IL-8, there is an increase in GRβ, which subsequently leads to a decrease in corticosteroid-induced apoptosis [192]. Similarly, an increase in GRβ: GRα ratio in male fetuses from pregnancies complicated by asthma could potentially allow these male fetuses to continue growing despite a high glucocorticoid environment, by upregulating genes related to growth and preventing apoptosis [187]. Unlike male fetuses, female fetuses have reduced growth in high glucocorticoid environments [174]. In SGA female placentae there is a decrease in the nuclear expression of GRβ [187]. This change is coupled with an increase in the nuclear expression of GRα-D3 and an increase in the cytoplasmic expression of GRα-C [187]. The interaction of GRα-A with GRα-C or GRα-D3 could contribute to maintaining glucocorticoid sensitivity in female placentae [187], potentially inducing higher rates of apoptosis, thereby contributing to reduced fetal growth. In preterm placentae, males displayed elevated cytoplasmic levels of GRα-C and GR-A, while female placentae showed increased nuclear levels of GRα-C compared to term placentae [188]. GRα-C is recognized as a powerful activator of glucocorticoid-triggered apoptosis, a process that includes the stimulation of the mitochondrial BIM pathway, consequently leading to the activation of caspases 9 and 3, resulting in cell death [193]. The placenta of preterm pregnancies has been shown to exhibit an increased expression of BIM and BAD [194]. This upregulation of GRα-C might be crucial in understanding the pathophysiology of preterm birth. It's possible that an escalation in glucocorticoid-induced cell death, mediated by GRα-C, could be a precursor to preterm labour [174]. These findings suggest that in a high glucocorticoid environment, the responsiveness to glucocorticoids hinges on the presence or absence of different GR variants, as well as the concurrent expression and interaction between different GR isoforms within placental tissue.
10. Conclusions
Glucocorticoids play a central role in regulating female reproduction and can act as a mediator of immune suppression or an activator of inflammation depending on the GR isoform profile. GR has been identified in many tissues and cell types of the reproductive tract and has a central role in pregnancy. However. the lack of data identifying the expression of different GR isoforms and their role in regulating the reproductive cycle is an area that requires further investigation.
Author Contributions
Conceptualization, V.C.; writing—original draft preparation, S.B.; writing—review and editing, S.B., V.C., J.L., J.C.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. Journal of Biological Chemistry. 2011, 286, 3177–3184. [Google Scholar] [CrossRef]
- Timmermans S, Souffriau J, Libert C. A general introduction to glucocorticoid biology. Frontiers in immunology. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues in clinical neuroscience 2022.
- Aguilera, G. Regulation of pituitary ACTH secretion during chronic stress. Frontiers in neuroendocrinology. 1994, 15, 321–350. [Google Scholar] [CrossRef]
- Sawchenko, P. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain research. 1987, 403, 213–224. [Google Scholar] [CrossRef]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrine reviews. 1984, 5, 25–44. [Google Scholar] [CrossRef]
- Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy, Asthma & Clinical Immunology. 2013, 9, 1–25.
- Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends in Endocrinology & Metabolism. 2013, 24, 109–119. [Google Scholar]
- Cruz-Topete D, Cidlowski JA. One hormone, two actions: anti-and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. 2015, 22, 20–32. [Google Scholar] [CrossRef]
- Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Molecular and cellular endocrinology. 2007, 275, 30–42. [Google Scholar] [CrossRef]
- Besedovsky H, Del Rey A, Klusman I, Furukawa H, Arditi GM, Kabiersch A. Cytokines as modulators of the hypothalamus-pituitary-adrenal axis. The Journal of steroid biochemistry and molecular biology. 1991, 40, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Molecular and cellular endocrinology. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Galati A, Brown ES, Bove R, Vaidya A, Gelfand J. Glucocorticoids for therapeutic immunosuppression: Clinical pearls for the practicing neurologist. Journal of the Neurological Sciences. 2021, 430, 120004. [Google Scholar] [CrossRef]
- FAUCI AS, DALE DC, BALOW JE. Glucocorticosteroid therapy: mechanisms of action and clinical considerations. Annals of internal medicine. 1976, 84, 304–315. [Google Scholar] [CrossRef]
- Settipane GA, Pudupakkam R, McGowan JH. Corticosteroid effect on immunoglobulins. Journal of Allergy and Clinical Immunology. 1978, 62, 162–166. [Google Scholar] [CrossRef]
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010, 75, 1–12. [Google Scholar] [CrossRef]
- Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best practice & research Clinical endocrinology & metabolism. 2015, 29, 545–556. [Google Scholar]
- Lu NZ, Wardell SE, Burnstein KL, Defranco D, Fuller PJ, Giguere V, et al. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacological reviews. 2006, 58, 782–797. [Google Scholar] [CrossRef]
- Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocrine reviews. 2014, 35, 671–693. [Google Scholar] [CrossRef]
- Bamberger CM, Bamberger A-M, De Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. The Journal of clinical investigation. 1995, 95, 2435–2441. [Google Scholar] [CrossRef]
- Moalli PA, Pillay S, Krett NL, Rosen ST. Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer research. 1993, 53, 3877–3879. [Google Scholar]
- Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, et al. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985, 318, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Oakley RH, Sar M, Cidlowski JA. The Human Glucocorticoid Receptor β Isoform: Expression, biochemical properties, and putative function (∗). Journal of Biological Chemistry. 1996, 271, 9550–9559.
- Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) β has intrinsic, GRα-independent transcriptional activity. Biochemical and biophysical research communications. 2009, 381, 671–675. [Google Scholar] [CrossRef] [PubMed]
- He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human glucocorticoid receptor β regulates gluconeogenesis and inflammation in mouse liver. Molecular and cellular biology. 2016, 36, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. The Journal of Clinical Endocrinology & Metabolism. 1999, 84, 4283–4286. [Google Scholar]
- Ray DW, Davis JR, White A, Clark AJ. Glucocorticoid receptor structure and function in glucocorticoid-resistant small cell lung carcinoma cells. Cancer research. 1996, 56, 3276–3280. [Google Scholar]
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009, 324, 407–410. [Google Scholar] [CrossRef]
- Thomas-Chollier M, Watson LC, Cooper SB, Pufall MA, Liu JS, Borzym K, et al. A naturally occuring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proceedings of the National Academy of Sciences. 2013, 110, 17826–17831. [Google Scholar] [CrossRef]
- Morgan DJ, Poolman TM, Williamson AJ, Wang Z, Clark NR, Ma’ayan A, et al. Glucocorticoid receptor isoforms direct distinct mitochondrial programs to regulate ATP production. Scientific reports. 2016, 6, 26419. [Google Scholar] [CrossRef]
- Gaitan D, DeBold CR, Turney MK, Zhou P, Orth DN, Kovacs WJ. Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Molecular endocrinology. 1995, 9, 1193–1201. [Google Scholar]
- Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Molecular cell. 2005, 18, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy S, Cidlowski JA. Corticosteroids: mechanisms of action in health and disease. Rheumatic Disease Clinics. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Hammond, GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. The Journal of endocrinology. 2016, 230, R13. [Google Scholar] [CrossRef]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocrine reviews. 1997, 18, 306–360. [Google Scholar]
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011, 145, 224–241. [Google Scholar] [CrossRef]
- Rogatsky I, Ivashkiv L. Glucocorticoid modulation of cytokine signaling. Tissue antigens. 2006, 68, 1–12. [Google Scholar] [CrossRef]
- Pantaleon M, Steane SE, McMahon K, Cuffe JS, Moritz KM. Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Scientific Reports. 2017, 7, 1–13. [Google Scholar]
- Groeneweg FL, Karst H, de Kloet ER, Joëls M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Molecular and cellular endocrinology. 2012, 350, 299–309. [Google Scholar] [CrossRef]
- Samarasinghe RA, Witchell SF, DeFranco DB. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle. 2012, 11, 2819–2827. [Google Scholar] [CrossRef]
- Dagklis T, Ravanos K, Makedou K, Kourtis A, Rousso D. Common features and differences of the hypothalamic–pituitary–gonadal axis in male and female. Gynecological Endocrinology. 2015, 31, 14–17. [Google Scholar] [CrossRef]
- Gharib SD, Wierman ME, Shupnik MA, Chin WW. Molecular biology of the pituitary gonadotropins. Endocrine reviews. 1990, 11, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Richards JS, Pangas SA. The ovary: basic biology and clinical implications. The Journal of clinical investigation. 2010, 120, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Whirledge S, Cidlowski JA. A role for glucocorticoids in stress-impaired reproduction: beyond the hypothalamus and pituitary. Endocrinology. 2013, 154, 4450–4468. [Google Scholar] [CrossRef] [PubMed]
- Geraghty AC, Kaufer D. Glucocorticoid regulation of reproduction. Glucocorticoid Signaling: From Molecules to Mice to Man. 2015, 253–278.
- Whirledge S, Cidlowski JA. Glucocorticoids and reproduction: traffic control on the road to reproduction. Trends in Endocrinology & Metabolism. 2017, 28, 399–415. [Google Scholar]
- Poisson M, Pertuiset B, Moguilewsky M, Magdelenat H, Martin P. Steroid receptors in the central nervous system. Implications in neurology. Revue neurologique. 1984, 140, 233–248. [Google Scholar]
- Chandran UR, Attardi B, Friedman R, Dong KW, Roberts JL, DeFranco DB. Glucocorticoid receptor-mediated repression of gonadotropin-releasing hormone promoter activity in GT1 hypothalamic cell lines. Endocrinology. 1994, 134, 1467–1474. [Google Scholar] [CrossRef]
- Gore AC, Attardi B, DeFranco DB. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Molecular and cellular endocrinology. 2006, 256, 40–48. [Google Scholar] [CrossRef]
- HYDE CL, CHILDS MORIARTY G, WAHL LM, NAOR Z, CATT KJ. Preparation of gonadotroph-enriched cell populations from adult rat anterior pituitary cells by centrifugal elutriation. Endocrinology. 1982, 111, 1421–1423. [Google Scholar] [CrossRef]
- Cheng KW, Leung PC. The expression, regulation and signal transduction pathways of the mammalian gonadotropin-releasing hormone receptor. Canadian journal of physiology and pharmacology. 2000, 78, 1029–1052. [Google Scholar] [CrossRef]
- Maya-Núñez G, Conn PM. Transcriptional regulation of the GnRH receptor gene by glucocorticoids. Molecular and cellular endocrinology. 2003, 200, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kilen SM, Szabo M, Strasser GA, McAndrews J, Ringstrom S, Schwartz N. Corticosterone selectively increases follicle-stimulating hormone beta-subunit messenger ribonucleic acid in primary anterior pituitary cell culture without affecting its half-life. Endocrinology. 1996, 137, 3802–3807. [Google Scholar] [CrossRef] [PubMed]
- Takumi K, Iijima N, Higo S, Ozawa H. Immunohistochemical analysis of the colocalization of corticotropin-releasing hormone receptor and glucocorticoid receptor in kisspeptin neurons in the hypothalamus of female rats. Neuroscience letters. 2012, 531, 40–45. [Google Scholar] [CrossRef]
- Luo E, Stephens SB, Chaing S, Munaganuru N, Kauffman AS, Breen KM. Corticosterone blocks ovarian cyclicity and the LH surge via decreased kisspeptin neuron activation in female mice. Endocrinology. 2016, 157, 1187–1199. [Google Scholar] [CrossRef]
- Ubuka T, Morgan K, Pawson AJ, Osugi T, Chowdhury VS, Minakata H, et al. Identification of human GnIH homologs, RFRP-1 and RFRP-3, and the cognate receptor, GPR147 in the human hypothalamic pituitary axis. PloS one. 2009, 4, e8400. [Google Scholar]
- Clarke IJ, Bartolini D, Conductier G, Henry BA. Stress increases gonadotropin inhibitory hormone cell activity and input to GnRH cells in ewes. Endocrinology. 2016, 157, 4339–4350. [Google Scholar] [CrossRef]
- Herrenkohl, LR. Prenatal stress reduces fertility and fecundity in female offspring. Science. 1979, 206, 1097–1099. [Google Scholar] [CrossRef]
- Carney E, Crissman J, Liberacki A, Clements C, Breslin W. Assessment of adult and neonatal reproductive parameters in Sprague-Dawley rats exposed to propylene glycol monomethyl ether vapors for two generations. Toxicological sciences: an official journal of the Society of Toxicology. 1999, 50, 249–258. [Google Scholar] [CrossRef]
- Crump C, Chevins P. Prenatal stress reduces fertility of male offspring in mice, without affecting their adult testosterone levels. Hormones and behavior. 1989, 23, 333–343. [Google Scholar] [CrossRef]
- De Rensis F, Scaramuzzi RJ. Heat stress and seasonal effects on reproduction in the dairy cow—a review. Theriogenology. 2003, 60, 1139–1151. [Google Scholar] [CrossRef]
- Schmid B, Tam-Dafond L, Jenni-Eiermann S, Arlettaz R, Schaub M, Jenni L. Modulation of the adrenocortical response to acute stress with respect to brood value, reproductive success and survival in the Eurasian hoopoe. Oecologia. 2013, 173, 33–44. [Google Scholar] [CrossRef]
- Lucas LD, French SS. Stress-induced tradeoffs in a free-living lizard across a variable landscape: consequences for individuals and populations. PloS one. 2012, 7, e49895. [Google Scholar]
- Akers SW, Mitchell CA. Seismic stress effects on reproductive structures of tomato, potato, and marigold. HortScience. 1985, 20, 684–686. [Google Scholar] [CrossRef]
- Hobel CJ, Goldstein A, Barrett ES. Psychosocial stress and pregnancy outcome. Clinical obstetrics and gynecology. 2008, 51, 333–348. [Google Scholar] [CrossRef]
- Mulder EJ, De Medina PR, Huizink AC, Van den Bergh BR, Buitelaar JK, Visser GH. Prenatal maternal stress: effects on pregnancy and the (unborn) child. Early human development. 2002, 70, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Beals KA, Meyer NL. Female athlete triad update. Clinics in sports medicine. 2007, 26, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Stafford, DE. Altered hypothalamic-pituitary-ovarian axis function in young female athletes: implications and recommendations for management. Treatments in Endocrinology. 2005, 4, 147–154. [Google Scholar] [CrossRef]
- Marilus R, Dickerman Z, Kaufman H, Varsano I, Laron Z. Addison's disease associated with precocious sexual development in a boy. Acta Pædiatrica. 1981, 70, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Zadik Z, Cooper M, Chen M, Stern N. Cushing's disease presenting as pubertal arrest. The Journal of Pediatric Endocrinology. 1993, 6, 201–204. [Google Scholar]
- Kinouchi R, Matsuzaki T, Iwasa T, Gereltsetseg G, Nakazawa H, Kunimi K, et al. Prepubertal exposure to glucocorticoid delays puberty independent of the hypothalamic Kiss1-GnRH system in female rats. International Journal of Developmental Neuroscience. 2012, 30, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Killian D, Kiesling D, Wulff F, Stewart A. Effects of adrenalectomy and glucocorticoids on puberty in gilts reared in confinement. Journal of Animal Science. 1987, 64, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Smith JT, Waddell BJ. Increased fetal glucocorticoid exposure delays puberty onset in postnatal life. Endocrinology. 2000, 141, 2422–2428. [Google Scholar] [CrossRef] [PubMed]
- Politch JA, Herrenkohl LR. Prenatal ACTH and corticosterone: effects on reproduction in male mice. Physiology & behavior. 1984, 32, 135–137. [Google Scholar]
- Politch JA, Herrenkohl LR. Effects of prenatal stress on reproduction in male and female mice. Physiology & behavior. 1984, 32, 95–99. [Google Scholar]
- Benešová O, Pavlik A. Perinatal treatment with glucocorticoids and the risk of maldevelopment of the brain. Neuropharmacology. 1989, 28, 89–97. [Google Scholar] [CrossRef]
- Omura T, Morohashi K-i. Gene regulation of steroidogenesis. The Journal of steroid biochemistry and molecular biology. 1995, 53, 19–25. [Google Scholar] [CrossRef]
- Jeon H, Choi Y, Brännström M, Akin J, Curry T, Jo M. Cortisol/glucocorticoid receptor: a critical mediator of the ovulatory process and luteinization in human periovulatory follicles. Human Reproduction. 2023, 38, 671–685. [Google Scholar] [CrossRef]
- Amin M, Simerman A, Cho M, Singh P, Briton-Jones C, Hill D, et al. 21-Hydroxylase-derived steroids in follicles of nonobese women undergoing ovarian stimulation for in vitro fertilization (IVF) positively correlate with lipid content of luteinized granulosa cells (LGCs) as a source of cholesterol for steroid synthesis. The Journal of Clinical Endocrinology & Metabolism. 2014, 99, 1299–1306. [Google Scholar]
- Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 1 lβ-hydroxysteroid dehydrogenase type 2 enzyme. Molecular and cellular endocrinology. 1994, 105, R11–R7. [Google Scholar] [CrossRef]
- Benediktsson R, Yau J, Low S, Brett L, Cooke B, Edwards C, et al. 11β-Hydroxysteroid dehydrogenase in the rat ovary: high expression in the oocyte. Journal of Endocrinology. 1992, 135, 53. [Google Scholar] [CrossRef] [PubMed]
- Condon J, Ricketts M, Whorwood C, Stewart P. Ontogeny and sexual dimorphic expression of mouse type 2 11β-hydroxysteroid dehydrogenase. Molecular and cellular endocrinology. 1997, 127, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kushnir MM, Naessen T, Kirilovas D, Chaika A, Nosenko J, Mogilevkina I, et al. Steroid profiles in ovarian follicular fluid from regularly menstruating women and women after ovarian stimulation. Clinical chemistry. 2009, 55, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Chapman K, Holmes M, Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiological reviews. 2013, 93, 1139–1206. [Google Scholar] [CrossRef]
- Tetsuka M, Thomas F, Thomas M, Anderson R, Mason J, Hillier S. Differential expression of messenger ribonucleic acids encoding 11beta-hydroxysteroid dehydrogenase types 1 and 2 in human granulosa cells. The Journal of clinical endocrinology and metabolism. 1997, 82, 2006–2009. [Google Scholar]
- Poulsen L, Bøtkjær J, Østrup O, Petersen K, Andersen CY, Grøndahl M, et al. Two waves of transcriptomic changes in periovulatory human granulosa cells. Human Reproduction. 2020, 35, 1230–1245. [Google Scholar] [CrossRef]
- Fateh M, Ben-Rafael Z, Benadiva CA, Mastroianni Jr L, Flickinger GL. Cortisol levels in human follicular fluid. Fertility and sterility. 1989, 51, 538–541. [Google Scholar] [CrossRef]
- Nteeba J, Sanz-Fernandez MV, Rhoads RP, Baumgard LH, Ross JW, Keating AF. Heat stress alters ovarian insulin-mediated phosphatidylinositol-3 kinase and steroidogenic signaling in gilt ovaries. Biology of Reproduction. 2015, 92, 148–1. [Google Scholar]
- Gosden R, Hunter R, Telfer E, Torrance C, Brown N. Physiological factors underlying the formation of ovarian follicular fluid. Reproduction. 1988, 82, 813–825. [Google Scholar] [CrossRef]
- Fortune, J. Ovarian follicular growth and development in mammals. Biology of reproduction. 1994, 50, 225–232. [Google Scholar] [CrossRef]
- Gao Y, Chen F, Kong Q-Q, Ning S-F, Yuan H-J, Lian H-Y, et al. Stresses on female mice impair oocyte developmental potential: effects of stress severity and duration on oocytes at the growing follicle stage. Reproductive sciences. 2016, 23, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Yuan H-J, Han X, He N, Wang G-L, Gong S, Lin J, et al. Glucocorticoids impair oocyte developmental potential by triggering apoptosis of ovarian cells via activating the Fas system. Scientific reports. 2016, 6, 1–12. [Google Scholar]
- Zhang S-Y, Wang J-Z, Li J-J, Wei D-L, Sui H-S, Zhang Z-H, et al. Maternal restraint stress diminishes the developmental potential of oocytes. Biology of Reproduction. 2011, 84, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Andersen, CY. Effect of glucocorticoids on spontaneous and follicle-stimulating hormone induced oocyte maturation in mouse oocytes during culture. The Journal of Steroid Biochemistry and Molecular Biology. 2003, 85((2-5)), 423–427. [Google Scholar] [CrossRef]
- Van Merris V, Van Wemmel K, Cortvrindt R. In vitro effects of dexamethasone on mouse ovarian function and pre-implantation embryo development. Reproductive Toxicology. 2007, 23, 32–41. [Google Scholar] [CrossRef]
- Gong S, Sun G-Y, Zhang M, Yuan H-J, Zhu S, Jiao G-Z, et al. Mechanisms for the species difference between mouse and pig oocytes in their sensitivity to glucorticoids. Biology of Reproduction. 2017, 96, 1019–1030. [Google Scholar] [CrossRef]
- Yang J-G, Chen W-Y, Li PS. Effects of glucocorticoids on maturation of pig oocytes and their subsequent fertilizing capacity in vitro. Biology of reproduction. 1999, 60, 929–936. [Google Scholar] [CrossRef]
- Scarlet D, Ille N, Ertl R, Alves B, Gastal G, Paiva S, et al. Glucocorticoid metabolism in equine follicles and oocytes. Domestic animal endocrinology. 2017, 59, 11–22. [CrossRef]
- Yuan X-H, Yang B-Q, Hu Y, Fan Y-Y, Zhang L-X, Zhou J-C, et al. Dexamethasone altered steroidogenesis and changed redox status of granulosa cells. Endocrine. 2014, 47, 639–647. [CrossRef]
- Huang T-J, Shirley Li P. Dexamethasone inhibits luteinizing hormone-induced synthesis of steroidogenic acute regulatory protein in cultured rat preovulatory follicles. Biology of reproduction. 2001, 64, 163–170. [Google Scholar] [CrossRef]
- Michael A, Pester L, Curtis P, Shaw R, Edwards C, Cooke B. Direct inhibition of ovarian steroidogenesis by Cortisol and the modulatory role of 11β-hydroxysteroid dehydrogenase. Clinical endocrinology. 1993, 38, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Joseph DN, Whirledge S. Stress and the HPA axis: balancing homeostasis and fertility. International journal of molecular sciences. 2017, 18, 2224. [Google Scholar] [CrossRef]
- Poulain M, Frydman N, Duquenne C, N′ Tumba-Byn T, Benachi A, Habert R, et al. Dexamethasone induces germ cell apoptosis in the human fetal ovary. The Journal of Clinical Endocrinology & Metabolism. 2012, 97, E1890–E7. [Google Scholar]
- Sasson R, Amsterdam A. Pleiotropic anti-apoptotic activity of glucocorticoids in ovarian follicular cells. Biochemical pharmacology. 2003, 66, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Komiyama J, Nishimura R, Lee H-Y, Sakumoto R, Tetsuka M, Acosta TJ, et al. Cortisol is a suppressor of apoptosis in bovine corpus luteum. Biology of reproduction. 2008, 78, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Rae MT, Price D, Harlow CR, Critchley HO, Hillier SG. Glucocorticoid receptor-mediated regulation of MMP9 gene expression in human ovarian surface epithelial cells. Fertility and sterility. 2009, 92, 703–708. [Google Scholar] [CrossRef]
- Schreiber JR, Nakamura K, Erickson GF. Rat ovary glucocorticoid receptor: identification and characterization. Steroids. 1982, 39, 569–584. [Google Scholar] [CrossRef]
- Rae MT, Niven D, Critchley HO, Harlow CR, Hillier SG. Antiinflammatory steroid action in human ovarian surface epithelial cells. The Journal of Clinical Endocrinology & Metabolism. 2004, 89, 4538–4544. [Google Scholar]
- Tetsuka M, Milne M, Simpson G, Hillier S. Expression of 11β-hydroxysteroid dehydrogenase, glucocorticoid receptor, and mineralocorticoid receptor genes in rat ovary. Biology of Reproduction. 1999, 60, 330–335. [Google Scholar] [CrossRef]
- Michael AE, Papageorghiou AT. Potential significance of physiological and pharmacological glucocorticoids in early pregnancy. Human reproduction update. 2008, 14, 497–517. [Google Scholar] [CrossRef]
- Čikoš Š, Babeľová J, Špirková A, Burkuš J, Kovaříková V, Šefčíková Z, et al. Glucocorticoid receptor isoforms and effects of glucocorticoids in ovulated mouse oocytes and preimplantation embryos. Biology of Reproduction. 2019, 100, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Oakley RH, Webster JC, Sar M, Parker Jr CR, Cidlowski JA. Expression and subcellular distribution of the β-isoform of the human glucocorticoid receptor. Endocrinology. 1997, 138, 5028–5038. [Google Scholar] [CrossRef] [PubMed]
- Rhen T, Grissom S, Afshari C, Cidlowski JA. Dexamethasone blocks the rapid biological effects of 17β-estradiol in the rat uterus without antagonizing its global genomic actions. The FASEB journal. 2003, 17, 1849–1870. [Google Scholar] [CrossRef]
- Johnson D, Dey S. Role of histamine in implantation: dexamethasone inhibits estradiol-induced implantation in the rat. Biology of reproduction. 1980, 22, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Whirledge S, Xu X, Cidlowski JA. Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biology of reproduction. 2013, 89, 66–1. [Google Scholar]
- Dmowski W, Ding J, Shen J, Rana N, Fernandez B, Braun D. Apoptosis in endometrial glandular and stromal cells in women with and without endometriosis. Human reproduction. 2001, 16, 1802–1808. [Google Scholar] [CrossRef]
- Ponce C, Torres M, Galleguillos C, Sovino H, Boric MA, Fuentes A, et al. Nuclear factor κB pathway and interleukin-6 are affected in eutopic endometrium of women with endometriosis. Reproduction. 2009, 137, 727–737. [Google Scholar] [CrossRef]
- Meno C, Saijoh Y, Fujii H, Ikeda M, Yokoyama T, Yokoyama M, et al. Left–right asymmetric expression of the TGFβ-family member lefty in mouse embryos. Nature. 1996, 381, 151–155. [Google Scholar] [CrossRef]
- Tang M, Naidu D, Hearing P, Handwerger S, Tabibzadeh S. LEFTY, a member of the transforming growth factor-β superfamily, inhibits uterine stromal cell differentiation: a novel autocrine role. Endocrinology. 2010, 151, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa MK, Medrano TI, Lydon JP, Bigsby RM, Cooke PS. Maximal dexamethasone inhibition of luminal epithelial proliferation involves progesterone receptor (PR)-and non-PR-mediated mechanisms in neonatal mouse uterus. Biology of reproduction. 2015, 92, 122–1. [Google Scholar]
- Kuroda K, Venkatakrishnan R, Salker MS, Lucas ES, Shaheen F, Kuroda M, et al. Induction of 11β-HSD 1 and activation of distinct mineralocorticoid receptor-and glucocorticoid receptor-dependent gene networks in decidualizing human endometrial stromal cells. Molecular endocrinology. 2013, 27, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang J, Dunk C, Croy AB, Lye SJ. To serve and to protect: the role of decidual innate immune cells on human pregnancy. Cell and tissue research. 2016, 363, 249–265. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo AS, Schumacher A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology. 2016, 148, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Vinketova K, Mourdjeva M, Oreshkova T. Human decidual stromal cells as a component of the implantation niche and a modulator of maternal immunity. Journal of pregnancy. 2016, 2016. [Google Scholar]
- Whirledge SD, Oakley RH, Myers PH, Lydon JP, DeMayo F, Cidlowski JA. Uterine glucocorticoid receptors are critical for fertility in mice through control of embryo implantation and decidualization. Proceedings of the National Academy of Sciences. 2015, 112, 15166–15171. [Google Scholar] [CrossRef] [PubMed]
- Robertson SA, Jin M, Yu D, Moldenhauer LM, Davies MJ, Hull ML, et al. Corticosteroid therapy in assisted reproduction–immune suppression is a faulty premise. Human reproduction. 2016, 31, 2164–2173. [Google Scholar] [CrossRef]
- Mori M, Bogdan A, Balassa T, Csabai T, Szekeres-Bartho J, editors. The decidua—the maternal bed embracing the embryo—maintains the pregnancy. Seminars in immunopathology; 2016: Springer.
- Bartmann C, Segerer SE, Rieger L, Kapp M, Sütterlin M, Kämmerer U. Quantification of the predominant immune cell populations in decidua throughout human pregnancy. American journal of reproductive immunology. 2014, 71, 109–119. [Google Scholar] [CrossRef]
- Henderson TA, Saunders PT, Moffett-King A, Groome NP, Critchley HO. Steroid receptor expression in uterine natural killer cells. The Journal of Clinical Endocrinology & Metabolism. 2003, 88, 440–449. [Google Scholar]
- Quenby S, Kalumbi C, Bates M, Farquharson R, Vince G. Prednisolone reduces preconceptual endometrial natural killer cells in women with recurrent miscarriage. Fertility and sterility. 2005, 84, 980–984. [Google Scholar] [CrossRef]
- Porta C, Riboldi E, Ippolito A, Sica A, editors. Molecular and epigenetic basis of macrophage polarized activation. Seminars in immunology; 2015: Elsevier.
- Gustafsson C, Mjösberg J, Matussek A, Geffers R, Matthiesen L, Berg G, et al. Gene expression profiling of human decidual macrophages: evidence for immunosuppressive phenotype. PloS one. 2008, 3, e2078. [CrossRef]
- Heikkinen J, Möttönen M, Komi J, Alanen A, Lassila O. Phenotypic characterization of human decidual macrophages. Clinical & Experimental Immunology. 2003, 131, 498–505. [Google Scholar]
- Thiruchelvam U, Maybin JA, Armstrong GM, Greaves E, Saunders PT, Critchley HO. Cortisol regulates the paracrine action of macrophages by inducing vasoactive gene expression in endometrial cells. Journal of leukocyte biology. 2016, 99, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Horton J, Yamamoto S, Bryant-Greenwood G. Relaxin modulates proinflammatory cytokine secretion from human decidual macrophages. Biology of reproduction. 2011, 85, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V, et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. The Journal of clinical investigation. 2008, 118, 3954–3965. [Google Scholar]
- Matasić R, Dietz AB, Vuk-Pavlović S. Dexamethasone inhibits dendritic cell maturation by redirecting differentiation of a subset of cells. Journal of leukocyte biology. 1999, 66, 909–914. [Google Scholar] [CrossRef]
- Bigler MB, Egli SB, Hysek CM, Hoenger G, Schmied L, Baldin FS, et al. Stress-induced in vivo recruitment of human cytotoxic natural killer cells favors subsets with distinct receptor profiles and associates with increased epinephrine levels. PloS one. 2015, 10, e0145635. [Google Scholar]
- Busillo JM, Azzam KM, Cidlowski JA. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. Journal of Biological Chemistry. 2011, 286, 38703–38713. [Google Scholar] [CrossRef]
- Moutsatsou P, Sekeris CE. Steroid receptors in the uterus: implications in endometriosis. Annals of the New York Academy of Sciences. 2003, 997, 209–222. [Google Scholar] [CrossRef]
- Gupta S, Gyomorey S, Lye SJ, Gibb W, Challis JR. Effect of labor on glucocorticoid receptor (GRTotal, GRα, and GRβ) proteins in ovine intrauterine tissues. The Journal of the Society for Gynecologic Investigation: JSGI. 2003, 10, 136–144. [Google Scholar] [CrossRef]
- Thomson, M. The physiological roles of placental corticotropin releasing hormone in pregnancy and childbirth. Journal of physiology and biochemistry. 2013, 69, 559–573. [Google Scholar] [CrossRef]
- Linton E, Perkins A, Woods R, Eben F, Wolfe C, Behan D, et al. Corticotropin releasing hormone-binding protein (CRH-BP): plasma levels decrease during the third trimester of normal human pregnancy. The Journal of Clinical Endocrinology & Metabolism. 1993, 76, 260–262. [Google Scholar]
- Challis JR, Sloboda DM, Alfaidy N, Lye SJ, Gibb W, Patel FA, et al. Prostaglandins and mechanisms of preterm birth. Reproduction-Cambridge-. 2002, 124, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li X, Zhu P, Myatt L, Sun K. Roles of glucocorticoids in human parturition: a controversial fact? Placenta. 2014, 35, 291–296. [Google Scholar] [CrossRef]
- ANDERSON AB, Flint A, Turnbull A. Mechanism of action of glucocorticoids in induction of ovine parturition: effect on placental steroid metabolism. Journal of Endocrinology. 1975, 66, 61–70. [Google Scholar] [CrossRef]
- Challis JR, Lye SJ, Gibb W, Whittle W, Patel F, Alfaidy N. Understanding preterm labor. Annals of the New York Academy of Sciences. 2001, 943, 225–234. [Google Scholar] [CrossRef]
- Liggins G, Grieves S. Possible role for prostaglandin F2α in parturition in sheep. Nature. 1971, 232, 629–631. [Google Scholar] [CrossRef]
- Gyomorey S, Lye S, Gibb W, Challis J. Fetal-to-maternal progression of prostaglandin H2 synthase-2 expression in ovine intrauterine tissues during the course of labor. Biology of reproduction. 2000, 62, 797–805. [Google Scholar] [CrossRef]
- Challis JR, Matthews SG, Gibb W, Lye SJ. Endocrine and paracrine regulation of birth at term and preterm. Endocrine reviews. 2000, 21, 514–550. [Google Scholar]
- Wang W, Liu C, Sun K. Induction of amnion epithelial apoptosis by cortisol via tPA/plasmin system. Endocrinology. 2016, 157, 4487–4498. [Google Scholar] [CrossRef]
- Mi Y, Wang W, Zhang C, Liu C, Lu J, Li W, et al. Autophagic degradation of collagen 1A1 by cortisol in human amnion fibroblasts. Endocrinology. 2017, 158, 1005–1014. [Google Scholar] [CrossRef]
- Hampson V, Liu D, Billett E, Kirk S. Amniotic membrane collagen content and type distribution in women with preterm premature rupture of the membranes in pregnancy. BJOG: An International Journal of Obstetrics & Gynaecology. 1997, 104, 1087–1091. [Google Scholar]
- Wadhwa PD, Garite TJ, Porto M, Glynn L, Chicz-DeMet A, Dunkel-Schetter C, et al. Placental corticotropin-releasing hormone (CRH), spontaneous preterm birth, and fetal growth restriction: a prospective investigation. American journal of obstetrics and gynecology. 2004, 191, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Challis J, Matthews S, Van Meir C, Ramirez M. Current topic: the placental corticotrophin-releasing hormone-adrenocorticotrophin axis. Placenta. 1995, 16, 481–502. [Google Scholar] [CrossRef]
- Craft I, Day J, Brummer V, Horwell D, Morgan H. Betamethazone induction of labour. SAGE Publications; 1976.
- Nwosu UC, Wallach EE, Bolognese RJ. Initiation of labor by intraamniotic cortisol instillation in prolonged human pregnancy. Obstetrics and gynecology. 1976, 47, 137–142. [Google Scholar]
- Elliott JP, Radin TG. The effect of corticosteroid administration on uterine activity and preterm labor in high-order multiple gestations. Obstetrics & Gynecology. 1995, 85, 250–254. [Google Scholar]
- Alexander N, Rosenlöcher F, Stalder T, Linke J, Distler W, Morgner J, et al. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. The Journal of Clinical Endocrinology & Metabolism. 2012, 97, 3538–3544. [Google Scholar]
- Ballard PL, Ballard RA. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. American journal of obstetrics and gynecology. 1995, 173, 254–262. [Google Scholar] [CrossRef]
- Liggins, GC. Premature parturition after infusion of corticotrophin or cortisol into foetal lambs. The Journal of endocrinology. 1968, 42, 323–329. [Google Scholar] [CrossRef]
- García-Blanco A, Diago V, De La Cruz VS, Hervás D, Cháfer-Pericás C, Vento M. Can stress biomarkers predict preterm birth in women with threatened preterm labor? Psychoneuroendocrinology. 2017, 83, 19–24. [CrossRef]
- Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocrine reviews. 2006, 27, 141–169. [Google Scholar] [CrossRef]
- Class QA, Lichtenstein P, Långström N, D'onofrio BM. Timing of prenatal maternal exposure to severe life events and adverse pregnancy outcomes: a population study of 2.6 million pregnancies. Psychosomatic medicine. 2011, 73, 234. [Google Scholar] [CrossRef]
- Gluckman PD, Cutfield W, Hofman P, Hanson MA. The fetal, neonatal, and infant environments—the long-term consequences for disease risk. Early human development. 2005, 81, 51–59. [Google Scholar] [CrossRef]
- Barker, DJ. The developmental origins of chronic adult disease. Acta paediatrica. 2004, 93, 26–33. [Google Scholar] [CrossRef]
- Singh RR, Cuffe J, Moritz KM, editors. Short-and long-term effects of exposure to natural and synthetic glucocorticoids during development. Proceedings of the Australian Physiological Society; 2012.
- Erni K, Shaqiri-Emini L, La Marca R, Zimmermann R, Ehlert U. Psychobiological effects of prenatal glucocorticoid exposure in 10-year-old-children. Frontiers in psychiatry. 2012, 3, 104.
- Huh SY, Andrew R, Rich-Edwards JW, Kleinman KP, Seckl JR, Gillman MW. Association between umbilical cord glucocorticoids and blood pressure at age 3 years. BMC medicine. 2008, 6, 1–8. [Google Scholar]
- Cuffe J, Dickinson H, Simmons D, Moritz K. Sex specific changes in placental growth and MAPK following short term maternal dexamethasone exposure in the mouse. Placenta. 2011, 32, 981–989. [Google Scholar] [CrossRef] [PubMed]
- O'Connell BA, Moritz KM, Roberts CT, Walker DW, Dickinson H. The placental response to excess maternal glucocorticoid exposure differs between the male and female conceptus in spiny mice. Biology of reproduction. 2011, 85, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- O'Sullivan L, Cuffe JS, Paravicini TM, Campbell S, Dickinson H, Singh RR, et al. Prenatal exposure to dexamethasone in the mouse alters cardiac growth patterns and increases pulse pressure in aged male offspring. PLoS One. 2013, 8, e69149. [CrossRef] [PubMed]
- Braun T, Meng W, Shang H, Li S, Sloboda DM, Ehrlich L, et al. Early dexamethasone treatment induces placental apoptosis in sheep. Reproductive sciences. 2015, 22, 47–59. [Google Scholar] [CrossRef]
- Clifton V, Cuffe J, Moritz K, Cole T, Fuller P, Lu N, et al. The role of multiple placental glucocorticoid receptor isoforms in adapting to the maternal environment and regulating fetal growth. Placenta. 2017, 54, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Murphy VE, Gibson PG, Giles WB, Zakar T, Smith R, Bisits AM, et al. Maternal asthma is associated with reduced female fetal growth. American journal of respiratory and critical care medicine. 2003, 168, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Cuffe J, O'sullivan L, Simmons D, Anderson S, Moritz K. Maternal corticosterone exposure in the mouse has sex-specific effects on placental growth and mRNA expression. Endocrinology. 2012, 153, 5500–5511. [Google Scholar] [CrossRef] [PubMed]
- Bronson SL, Bale TL. Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology. 2014, 155, 2635–2646. [Google Scholar] [CrossRef] [PubMed]
- Cheong JN, Cuffe JS, Jefferies AJ, Anevska K, Moritz KM, Wlodek ME. Sex-specific metabolic outcomes in offspring of female rats born small or exposed to stress during pregnancy. Endocrinology. 2016, 157, 4104–4120. [Google Scholar] [CrossRef]
- Carpenter T, Grecian S, Reynolds R. Sex differences in early-life programming of the hypothalamic–pituitary–adrenal axis in humans suggest increased vulnerability in females: a systematic review. Journal of developmental origins of health and disease. 2017, 8, 244–255. [Google Scholar] [CrossRef]
- Clifton, V. Sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta. 2010, 31, S33–S9. [Google Scholar] [CrossRef]
- Mericq V, Medina P, Kakarieka E, Márquez L, Johnson M, Iniguez G. Differences in expression and activity of 11β-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. European Journal of Endocrinology. 2009, 161, 419–425. [Google Scholar] [CrossRef]
- Cuffe JS, Saif Z, Perkins AV, Moritz KM, Clifton VL. Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. Journal of Endocrinology. 2017, 234, 89–100. [Google Scholar] [CrossRef]
- Simcock G, Kildea S, Elgbeili G, Laplante D, Cobham V, King S. Prenatal maternal stress shapes children’s theory of mind: the QF2011 Queensland Flood Study. Journal of developmental origins of health and disease. 2017, 8, 483–492.
- Sandman CA, Glynn LM, Davis EP. Is there a viability–vulnerability tradeoff? Sex differences in fetal programming. Journal of psychosomatic research. 2013, 75, 327–335. [Google Scholar] [CrossRef]
- Braithwaite EC, Murphy SE, Ramchandani PG, Hill J. Associations between biological markers of prenatal stress and infant negative emotionality are specific to sex. Psychoneuroendocrinology. 2017, 86, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scott NM, Hodyl NA, Murphy VE, Osei-Kumah A, Wyper H, Hodgson DM, et al. Placental cytokine expression covaries with maternal asthma severity and fetal sex. The Journal of Immunology. 2009, 182, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Saif Z, Hodyl N, Hobbs E, Tuck A, Butler M, Osei-Kumah A, et al. The human placenta expresses multiple glucocorticoid receptor isoforms that are altered by fetal sex, growth restriction and maternal asthma. Placenta. 2014, 35, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Saif Z, Hodyl NA, Stark MJ, Fuller PJ, Cole T, Lu N, et al. Expression of eight glucocorticoid receptor isoforms in the human preterm placenta vary with fetal sex and birthweight. Placenta. 2015, 36, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Saif Z, Dyson RM, Palliser HK, Wright IM, Lu N, Clifton VL. Identification of eight different isoforms of the glucocorticoid receptor in guinea pig placenta: relationship to preterm delivery, sex and betamethasone exposure. PloS one. 2016, 11, e0148226. [Google Scholar]
- Clifton VL, McDonald M, Morrison JL, Holman SL, Lock MC, Saif Z, et al. Placental glucocorticoid receptor isoforms in a sheep model of maternal allergic asthma. Placenta. 2019, 83, 33–36. [Google Scholar] [CrossRef]
- Akison L, Andersen I, Kent N, Steane S, Saif Z, Clifton V, et al. Glucocorticoid receptor isoform expression profile in the rat placenta and fetal liver in pregnancies exposed to periconceptional alcohol. Alcoholism: Clinical and Experimental Research. 2018, 42(S1), 228A-A.
- Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, et al. High constitutive glucocorticoid receptor β in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. The Journal of experimental medicine. 2001, 193, 585–594. [Google Scholar] [CrossRef]
- Gruver-Yates AL, Cidlowski JA. Tissue-specific actions of glucocorticoids on apoptosis: a double-edged sword. Cells. 2013, 2, 202–223. [Google Scholar] [CrossRef]
- Whitehead C, Walker S, Lappas M, Tong S. Circulating RNA coding genes regulating apoptosis in maternal blood in severe early onset fetal growth restriction and pre-eclampsia. Journal of Perinatology. 2013, 33, 600–604. [Google Scholar] [CrossRef]
Figure 1.
Hypothalamic-Pituitary-Adrenal axis. Cells in the paraventricular nucleus produce CRH in response to physical or psychological stress. CRH binds to cells in the anterior pituitary gland to stimulate the production of ACTH which acts on the adrenal glands to increase the production of glucocorticoids which in turn regulates the stress response. Glucocorticoids can also exert its negative effect on CRH and ACTH via a negative feedback mechanism (-). PVN – Paraventricular nucleus, CRH – corticotrophin-releasing hormone, ACTH – adrenocorticotrophic hormone. Created on bio render. Adapted from [36].
Figure 1.
Hypothalamic-Pituitary-Adrenal axis. Cells in the paraventricular nucleus produce CRH in response to physical or psychological stress. CRH binds to cells in the anterior pituitary gland to stimulate the production of ACTH which acts on the adrenal glands to increase the production of glucocorticoids which in turn regulates the stress response. Glucocorticoids can also exert its negative effect on CRH and ACTH via a negative feedback mechanism (-). PVN – Paraventricular nucleus, CRH – corticotrophin-releasing hormone, ACTH – adrenocorticotrophic hormone. Created on bio render. Adapted from [36].
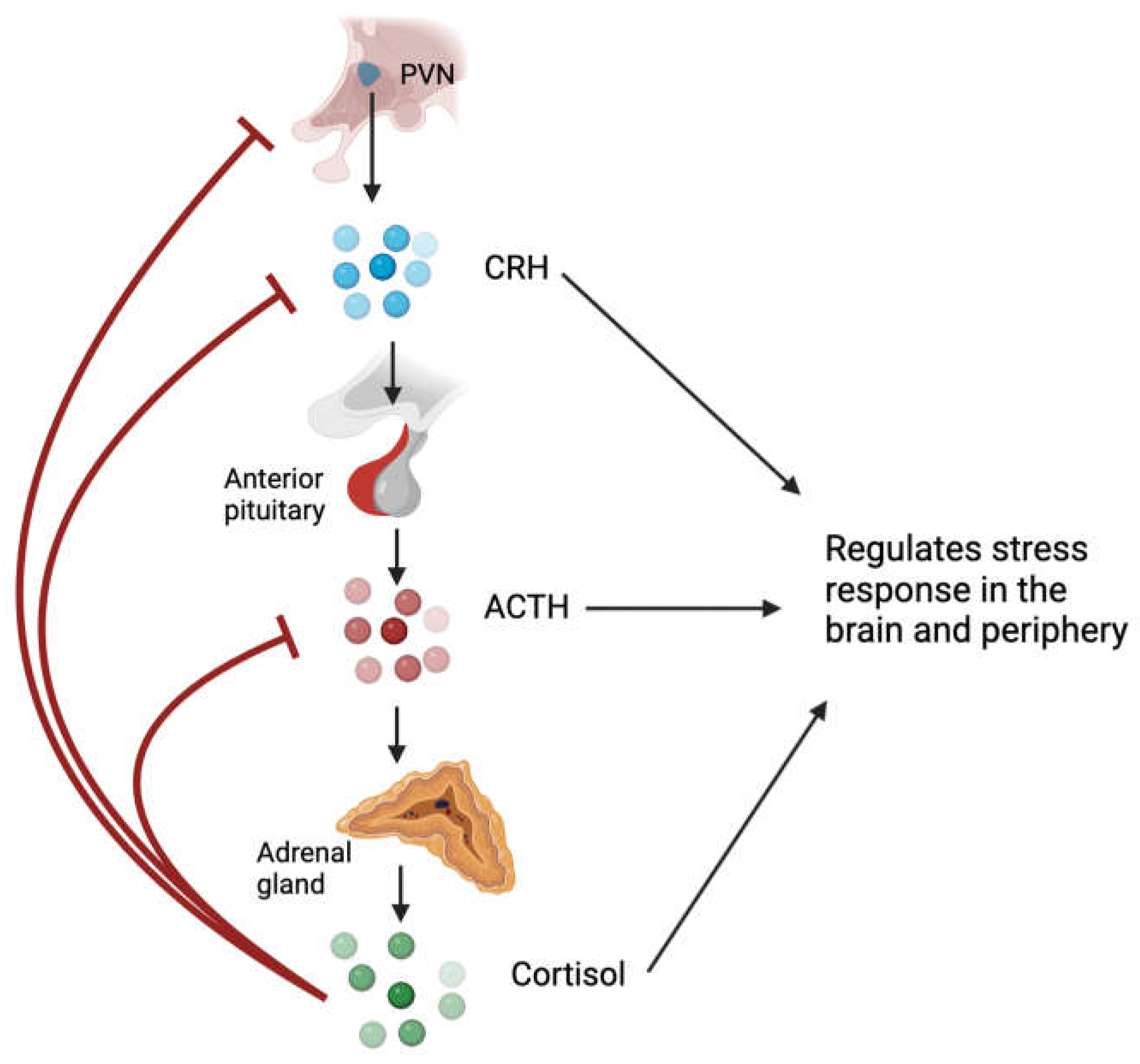
Figure 2.
Splice and translation initiation isoforms of the glucocorticoid receptor. Exons of the glucocorticoid receptor gene with the subsequent splice (left) and translation initiation (right) isoforms. AF-1 – transcriptional activation function 1, DBD – DNA binding domain, GR – glucocorticoid receptor, LBD – ligand-binding domain, NTD – N-terminal domain. Created on bio render. Adapted from Ramamoorthy & Cidlowski [33].
Figure 2.
Splice and translation initiation isoforms of the glucocorticoid receptor. Exons of the glucocorticoid receptor gene with the subsequent splice (left) and translation initiation (right) isoforms. AF-1 – transcriptional activation function 1, DBD – DNA binding domain, GR – glucocorticoid receptor, LBD – ligand-binding domain, NTD – N-terminal domain. Created on bio render. Adapted from Ramamoorthy & Cidlowski [33].
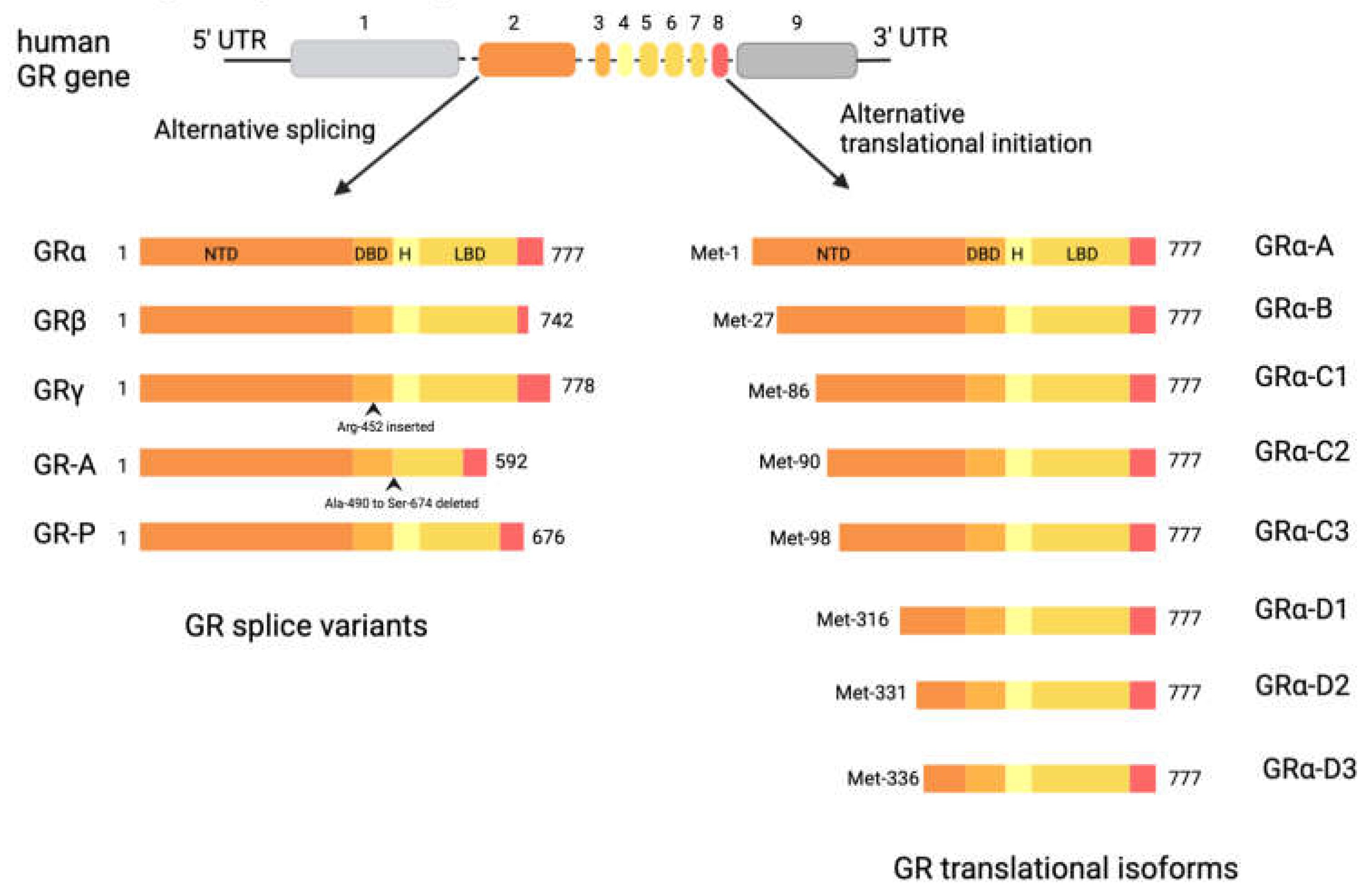
Figure 3.
Genomic action of Glucocorticoid receptor (GR). GR binds to glucocorticoids in the cytoplasm, leading to the dislocation of its chaperone protein complex and translocation into the nucleus. In the nucleus, GR can either directly induce or repress gene expression by binding to GRE or nGRE, respectively. It can also interact with other TFs by tethering or composite actions. GR- Glucocorticoid receptor, Hsp90 – Heat shock protein 90, Fkbp51 - FK506-binding protein 51, GRE – Glucocorticoid response element, nGRE- negative GRE, TF- Transcription factor. Created on bio render. Adapted from [52].
Figure 3.
Genomic action of Glucocorticoid receptor (GR). GR binds to glucocorticoids in the cytoplasm, leading to the dislocation of its chaperone protein complex and translocation into the nucleus. In the nucleus, GR can either directly induce or repress gene expression by binding to GRE or nGRE, respectively. It can also interact with other TFs by tethering or composite actions. GR- Glucocorticoid receptor, Hsp90 – Heat shock protein 90, Fkbp51 - FK506-binding protein 51, GRE – Glucocorticoid response element, nGRE- negative GRE, TF- Transcription factor. Created on bio render. Adapted from [52].
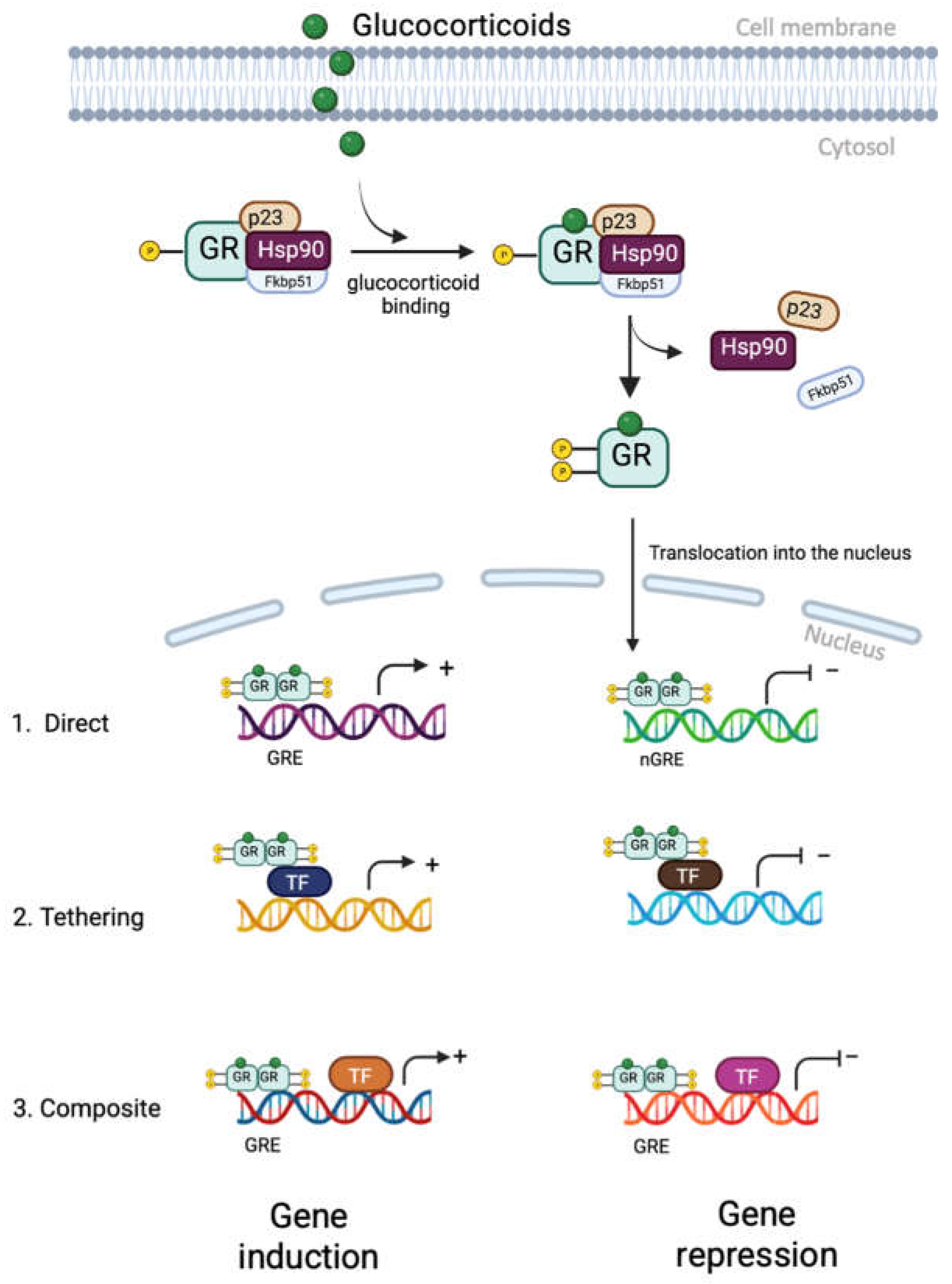
Figure 4.
Steroidogenesis pathway, illustrating the key factors involved in the conversion of cholesterol into progesterone and cortisol. STAR, steroidogenic acute regulatory protein; CYP11A1, cytochrome P450 family 11 subfamily A member 1; CYP17A1, cytochrome P450 family 17 subfamily A member 1; HSD3B, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase; CYP21A2, cytochrome P450 family 21 subfamily A member 2; CYP11B1, cytochrome P450 family 11 subfamily B member 1; HSD11B1, hydroxysteroid 11-beta dehydrogenase 1; HSD11B2, hydroxysteroid 11-beta dehydrogenase 2. Created on bio render. Adapted from [78].
Figure 4.
Steroidogenesis pathway, illustrating the key factors involved in the conversion of cholesterol into progesterone and cortisol. STAR, steroidogenic acute regulatory protein; CYP11A1, cytochrome P450 family 11 subfamily A member 1; CYP17A1, cytochrome P450 family 17 subfamily A member 1; HSD3B, hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase; CYP21A2, cytochrome P450 family 21 subfamily A member 2; CYP11B1, cytochrome P450 family 11 subfamily B member 1; HSD11B1, hydroxysteroid 11-beta dehydrogenase 1; HSD11B2, hydroxysteroid 11-beta dehydrogenase 2. Created on bio render. Adapted from [78].
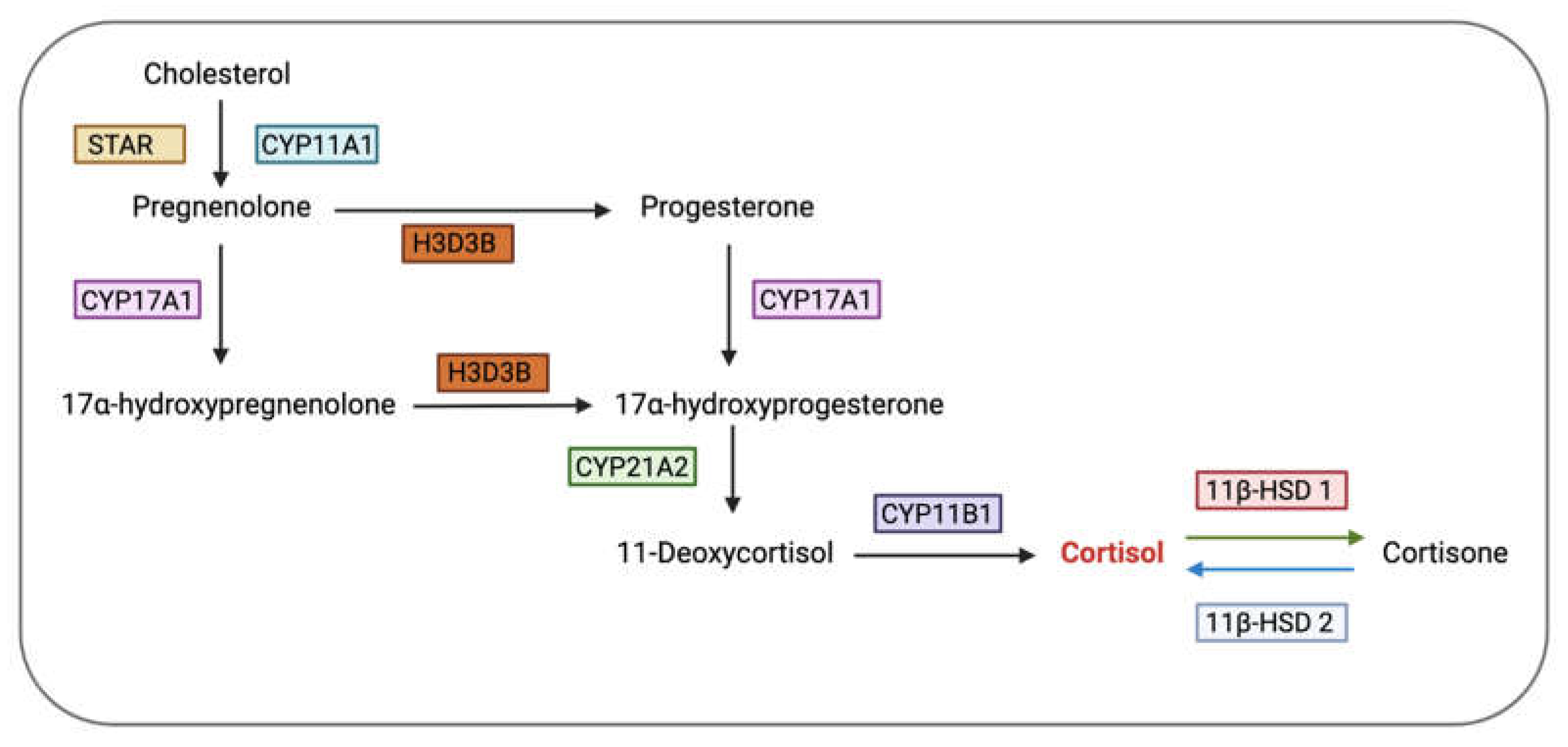
Figure 5.
Stress axis during pregnancy. Under stress, the maternal HPA axis produces cortisol which can act on the placenta to stimulate the production of pCRH. Most of the cortisol is metabolized by placental 11β-HSD 2, with 20% crossing over to the fetus. Increased levels of cortisol in the fetal compartment can over activate the fetal HPA leading to long-lasting effects on its growth and development. CRH- Corticotrophin releasing hormone, ACTH- Adrenocorticotropic hormone, 11β-HSD 2- Type 2 11-beta hydroxysteroid dehydrogenase, pCRH- placental CRH, HPA- Hypothalamus-pituitary-adrenal. Created on bio render.
Figure 5.
Stress axis during pregnancy. Under stress, the maternal HPA axis produces cortisol which can act on the placenta to stimulate the production of pCRH. Most of the cortisol is metabolized by placental 11β-HSD 2, with 20% crossing over to the fetus. Increased levels of cortisol in the fetal compartment can over activate the fetal HPA leading to long-lasting effects on its growth and development. CRH- Corticotrophin releasing hormone, ACTH- Adrenocorticotropic hormone, 11β-HSD 2- Type 2 11-beta hydroxysteroid dehydrogenase, pCRH- placental CRH, HPA- Hypothalamus-pituitary-adrenal. Created on bio render.
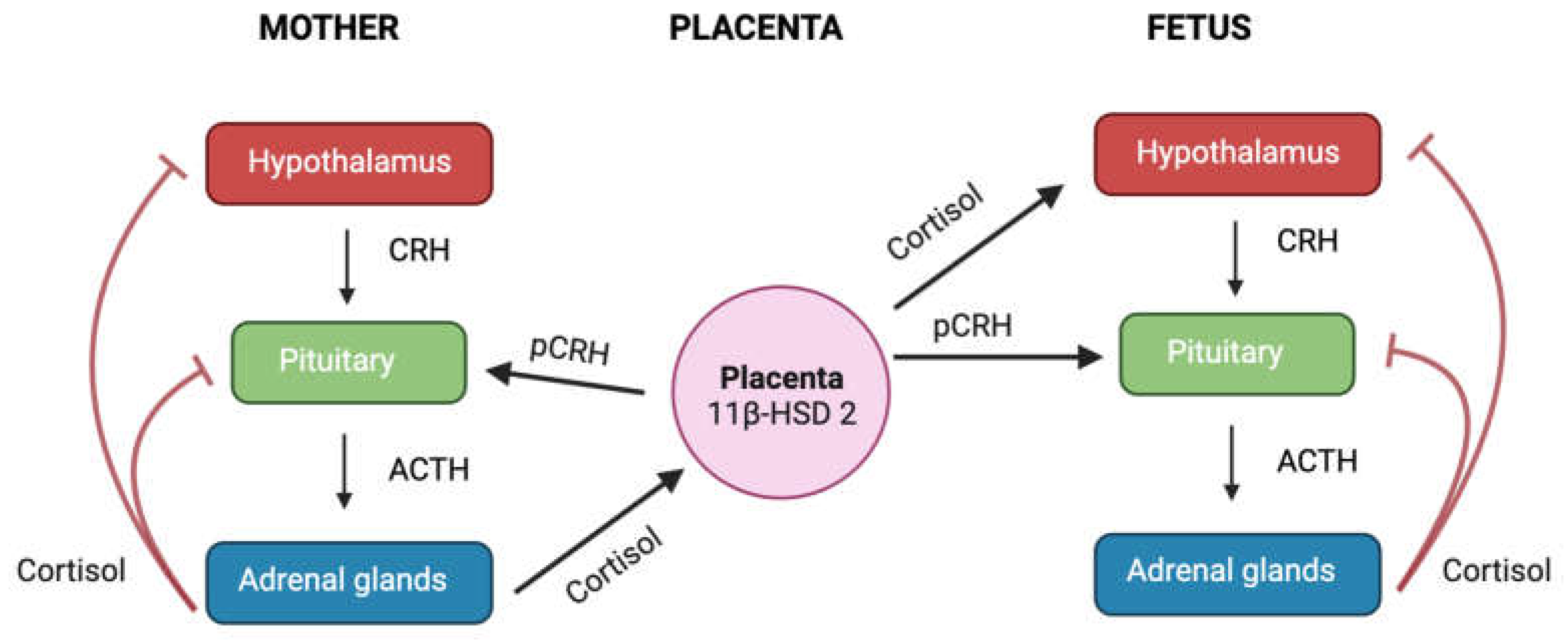
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



