
Submitted:
28 February 2024
Posted:
28 February 2024
You are already at the latest version
Abstract
The corpus luteum is a temporary endocrine gland formed in the ovary after ovulation, and it plays a critical role in reproductive processes. Tumors rely on the development of an adequate blood supply to ensure the delivery of nutrients and oxygen and the removal of waste products. While angiogenesis occurs in various physiological and pathological contexts, the corpus luteum and tumors share similarities in the signaling pathways that promote angiogenesis. In the corpus luteum and tumors, apoptosis plays a crucial role in controlling cell numbers and ensuring proper tissue development and function. Interestingly, there are similarities in the apoptotic-regulated signaling pathways involved in apoptosis between the corpus luteum and tumors. However, the regulation of apoptosis can differ due to their distinct physiological and pathological characteristics. Thus, we reviewed the biological events of the corpus luteum and tumors in similar microenvironments of angiogenesis and apoptosis for studying novel research methods.
Keywords:
ovary
; corpus luteum
; tumor
; angiogenesis
; apoptosis
1. Introduction
Angiogenesis and apoptosis are two fundamental biological processes that play crucial roles in developing, maintaining, and functioning various tissues and organs in humans and animals. These processes are also intimately involved in the pathophysiology of diseases, including ovaries and cancer [1,2,3,4]. The microenvironments in which the processes of angiogenesis and apoptosis occur share some similarities between the corpus luteum and tumors (Figure 1).
The angiogenic microenvironment is characterized by a complex interplay of pro-angiogenic and anti-angiogenic factors, signaling molecules, and cellular components. In both the corpus luteum and tumors, angiogenesis is initiated by a state of hypoxia, which releases pro-angiogenic factors that stimulate the formation of new blood vessels [2,5]. Also, the angiogenic microenvironment involves cellular components, including endothelial cells, pericytes, and immune cells. Endothelial cells play a central role in angiogenesis, forming the inner lining of blood vessels. Pericytes, which surround the endothelial cells, provide stability and regulate vessel maturation. Immune cells, such as macrophages and lymphocytes, can promote or inhibit angiogenesis depending on their phenotype and secreted factors [1,6].
Apoptosis, or programmed cell death, is a tightly regulated process essential for maintaining tissue homeostasis. Also, the microenvironment of apoptosis involves a complex network of signaling molecules, cellular components, and extracellular factors [7]. In both the corpus luteum and tumors, apoptosis is regulated to maintain tissue homeostasis and eliminate abnormal or unwanted cells [8,9]. The microenvironments in which apoptosis occurs share several similarities between the corpus luteum and tumors. These similarities involve cellular interactions, signaling molecules, and extracellular factors that govern the apoptotic process.
Therefore, we reviewed similar microenvironments and mechanisms of angiogenesis and apoptosis in the corpus luteum and tumor. Understanding these shared features can provide insights into the regulation and dysregulation of angiogenesis and apoptosis in both microenvironments for approaching a research topic from a novel viewpoint.
2. Angiogenesis in Corpus Luteum and Tumor
Angiogenesis is a complex biological process that involves the formation of new blood vessels from pre-existing ones. It plays a critical role in various physiological and pathological conditions, including the development of the corpus luteum and tumors [1,2]. Also, angiogenesis is a tightly regulated process involving the proliferation, migration, and differentiation of endothelial cells. Moreover, angiogenesis plays a pivotal role in physiological processes such as embryonic development, wound healing, and the menstrual cycle [10,11]. It can also contribute to the progression of diseases, particularly ovarian cancer [12].
Both in the corpus luteum and tumors, the balance between pro-angiogenic and anti-angiogenic factors is critical. In the corpus luteum, as it transitions from the early luteal phase to the mid-luteal phase, anti-angiogenic factors, such as thrombospondin-1 (TSP-1) and angiopoietin-2 (Ang-2), are upregulated. These factors help stabilize the vasculature and maintain a functional corpus luteum [13]. Similarly, in tumors, the expression of anti-angiogenic factors, including angiostatin and endostatin, can counteract the effects of pro-angiogenic factors. These anti-angiogenic factors inhibit endothelial cell proliferation and migration, maintaining a balance between angiogenic stimulators and inhibitors [14,15]. As mentioned above, the microenvironments of angiogenesis in the corpus luteum and tumors share several similarities in terms of cellular interactions, signaling molecules, and extracellular factors.
2.1. Angiogenesis in Corpus Luteum
In the corpus luteum, following ovulation, the ruptured follicle transforms into a structure called the corpus luteum, which requires angiogenesis to sustain its function [16]. Hypoxia within the developing corpus luteum triggers the secretion of pro-angiogenic factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). These factors promote endothelial cell proliferation, migration, and tube formation, leading to the formation of new blood vessels [5,17,18]. Similarly, in tumors, rapid growth leads to an inadequate blood supply, resulting in hypoxia. This hypoxic state triggers the release of pro-angiogenic factors, primarily VEGF, by tumor cells and stroma cells. The pro-angiogenic factors promote the recruitment of new blood vessels that supply oxygen and nutrients to the growing tumor [19].
2.2. Angiogenesis in Tumor
Tumors, characterized by uncontrolled cell growth and division, require a dedicated blood supply to sustain their growth and survival. Angiogenesis is a critical step in tumor microenvironment [20,21]. This process is regulated by a delicate balance between proangiogenic and antiangiogenic factors. Proangiogenic factors, such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF), are secreted by tumor cells and stromal cells in response to hypoxia and other stimuli. These factors promote the proliferation and migration of endothelial cells. They lead to the sprouting of new blood vessels toward the tumor [12,20,22]. New blood vessels are often disorganized and leaky, further contributing to the chaotic growth of the tumor.
2.3. Cellular Interaction
The angiogenic microenvironment in the corpus luteum and tumors involves interactions between various cell types, including endothelial cells, pericytes, immune cells, and stromal cells. Endothelial cells are the primary cellular component of blood vessels and play a central role in angiogenesis. In both the corpus luteum and tumors, endothelial cells are stimulated to proliferate, migrate, and form new blood vessels in response to pro-angiogenic signals. Pericytes, located around endothelial cells, provide structural support to blood vessels and regulate vessel stability and maturation. They interact with endothelial cells through cell-cell contacts and secrete factors that modulate angiogenesis [23,24]. Immune cells, such as macrophages and lymphocytes, are also present in the angiogenic microenvironments of both the corpus luteum and tumors. These immune cells can secrete pro-angiogenic factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), to promote angiogenesis [6,25,26,27]. Alternatively, they can release anti-angiogenic factors, such as thrombospondin-1 (TSP-1) and angiostatin, to inhibit angiogenesis [6,28].
2.4. Signaling Molecules
Various signaling molecules play crucial roles in regulating angiogenesis in the corpus luteum and tumors. These molecules include growth factors, cytokines, chemokines, and extracellular matrix (ECM) components. In the corpus luteum, angiogenesis is tightly regulated by the balance between pro-angiogenic and anti-angiogenic factors. Pro-angiogenic factors, such as VEGF, FGF, and angiopoietin, are secreted by luteal cells and promote endothelial cell proliferation, migration, and tube formation. These factors act through specific receptors on endothelial cells, initiating intracellular signaling pathways that drive angiogenesis [17,18,29]. Similarly, in tumors, pro-angiogenic factors are released by tumor cells and stromal cells. VEGF, in particular, is a key regulator of tumor angiogenesis. It stimulates endothelial cell proliferation, migration, and the formation of new blood vessels [30]. Other pro-angiogenic factors, such as FGF, PDGF, and angiopoietin, also contribute to tumor angiogenesis [12,18].
2.5. Extracellular Factors
The extracellular matrix (ECM) is a complex network of proteins and proteoglycans that provides structural support to tissues and regulates cellular functions. In both the corpus luteum and tumors, the ECM plays a crucial role in modulating angiogenesis [20]. ECM proteins, such as fibronectin, collagen, laminin, and hyaluronic acid, are present in the angiogenic microenvironments of both the corpus luteum and tumors. These proteins interact with endothelial cells and other cell types, influencing cell adhesion, migration, and signaling [31,32]. The ECM also acts as a reservoir for growth factors and cytokines, sequestering and releasing them to regulate angiogenesis. Enzymes involved in ECM remodeling, such as matrix metalloproteinases (MMPs), are upregulated during angiogenesis [33,34].
2.6. Cytokines and Chemokines
Cytokines and chemokines are small signaling molecules that regulate cell communication and immune responses. They can also modulate angiogenesis by influencing endothelial cell behavior and promoting the recruitment of immune cells [35]. In the corpus luteum, various cytokines and chemokines are involved in angiogenesis. Interleukin-8 (IL-8), for instance, promotes angiogenesis by stimulating endothelial cell migration and proliferation [36,37,38]. Additionally, tumor necrosis factor-alpha (TNF-α) and transforming growth factor-beta (TGF-β) can induce the expression of pro-angiogenic factors and contribute to angiogenesis in the corpus luteum [20,39]. Similarly, in tumors, cytokines and chemokines play important roles in angiogenesis. Interleukin-6 (IL-6), interleukin-1 (IL-1), and TNF-α, among others, are produced by tumor cells and stromal cells. They can stimulate endothelial cell proliferation and migration, promote the production of pro-angiogenic factors, and attract immune cells to the tumor microenvironment [26,40].
2.7. Signaling Pathways
2.7.1. Vascular Endothelial Growth Factor (VEGF) Signaling Pathway
The VEGF pathway is one of the central signaling pathways involved in angiogenesis in both the corpus luteum and tumors. VEGF is a potent pro-angiogenic factor that stimulates endothelial cell proliferation, migration, and survival [41,42]. In the corpus luteum, luteal cells produce VEGF in response to luteinizing hormone (LH) stimulation [1,43]. VEGF binds to its receptors, particularly VEGFR-2 (also known as Flk-1), on endothelial cells, triggering downstream signaling events [30,44]. This activation leads to the activation of phospholipase C gamma (PLCγ), which in turn generates inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 induces calcium release from the endoplasmic reticulum, leading to the activation of protein kinase C (PKC). DAG, together with PKC, stimulates mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways [10,30,45]. The MAPK includes the extracellular signal-regulated kinase (ERK) cascade, promoting endothelial cell proliferation and migration [46].
Furthermore, VEGF also activates the PI3K pathway, leading to the activation of Akt (also known as protein kinase B). Akt promotes endothelial cell survival and migration by regulating various downstream effectors, including the mammalian target of rapamycin (mTOR) pathway [44,47]. In tumors, tumor cells and stromal cells secrete VEGF to promote angiogenesis within the tumor microenvironment [48]. The binding of VEGF to its receptors on endothelial cells activates similar downstream signaling pathways involving PLCγ, IP3, DAG, PKC, MAPK, and PI3K. These signaling cascades promote endothelial cell proliferation, migration, and the formation of new blood vessels to support tumor growth.
2.7.2. Fibroblast Growth Factor (FGF) Signaling Pathway
The FGF signaling pathway plays a critical role in angiogenesis by stimulating endothelial cell proliferation, migration, and tube formation. FGFs are a family of growth factors that bind to FGF receptors (FGFRs) on endothelial cells, initiating intracellular signaling cascades [49]. In the corpus luteum, FGFs are expressed by luteal cells and stromal cells [18,50]. The binding of FGFs to FGFRs on endothelial cells activates the Ras-MAPK pathway, leading to endothelial cell proliferation. The activation of the Ras-MAPK pathway results in the phosphorylation and activation of downstream effectors, including the ERK cascade. This pathway plays a crucial role in endothelial cell proliferation, migration, and survival, contributing to angiogenesis in the corpus luteum [51,52]. Additionally, FGF signaling activates the PI3K-Akt pathway, promoting endothelial cell survival, migration, and tube formation. Akt, a key mediator of this pathway, regulates multiple downstream effectors involved in angiogenesis, such as mTOR and endothelial nitric oxide synthase (eNOS) [47]. Similarly, tumor cells and stromal cells secrete FGFs to induce tumor angiogenesis. The binding of FGFs to FGFRs on endothelial cells activates Ras-MAPK and PI3K-Akt pathways, leading to endothelial cell proliferation, survival, and migration [10].
2.7.3. Platelet-Derived Growth Factor (PDGF) Signaling Pathway
The PDGF signaling pathway is involved in angiogenesis by promoting endothelial cell recruitment and pericyte stabilization of newly formed blood vessels [12,23]. Interestingly, there are similarities in PDGF signaling pathways between the angiogenesis of the corpus luteum and tumors. PDGFs are a family of growth factors that consist of five isoforms: PDGF-AA, -BB, -AB, -CC, and -DD. These isoforms bind to specific PDGF receptors, primarily PDGFR-β. The binding of PDGF ligands to their receptors initiates downstream signaling cascades that regulate angiogenesis [53,54,55]. The corpus luteum and tumor express PDGF ligands and receptors [39,55]. The binding of PDGF ligands to PDGFRs leads to receptor dimerization and activation. PDGF-α ligands primarily bind to PDGFR-α, while PDGF-β ligands can activate both PDGFR-α and PDGFR-β. Dimerization of PDGFRs results in autophosphorylation of specific tyrosine residues within the receptor cytoplasmic domain. This autophosphorylation creates docking sites for various intracellular signaling molecules, initiating downstream signaling events [56,57,58].
The PI3K pathway is a crucial downstream signaling pathway activated by PDGF in both the corpus luteum and tumors. The binding of PDGF ligands to PDGFRs leads to the recruitment and activation of PI3K. Activated PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to generate phosphatidylinositol 3,4,5,-trisphosphate (PIP3). PIP3 acts as a second messenger and recruits proteins containing pleckstrin homology (PH) domains, such as Akt, to the plasma membrane [53,57]. Akt is subsequently phosphorylated and activated by phosphoinositide-dependent kinase 1 (PDK1) and the mammalian target of rapamycin complex 2 (mTORC2). Activated Akt promotes cell survival, migration, and proliferation, contributing to angiogenesis [59,60].
The Ras-MAPK pathway is another important downstream signaling pathway activated by PDGF in both the corpus luteum and tumors. Upon PDGF binding, activated PDGFRs recruit and activate Ras guanine nucleotide exchange factors (GEFs), leading to Ras activation. Activated Ras initiates a cascade of phosphorylation events, culminating in the activation of extracellular signal-regulated kinases (ERKs) in the MAPK pathway. Activated ERKs translocate to the nucleus and phosphorylate various transcription factors, resulting in the expression of genes involved in endothelial cell proliferation, migration, and angiogenesis [54,57].
3. Apoptosis in Corpus Luteum and Tumor
The process of apoptosis, or programmed cell death, serves as a crucial mechanism for maintaining tissue homeostasis. Apoptosis eliminates unwanted or damaged cells, preventing their accumulation and potentially harmful effects. Dysregulation of apoptosis can result in various diseases [7]. In the corpus luteum, apoptosis occurs in a tightly regulated manner and is necessary for the regression of the structure. The lifespan of the corpus luteum is primarily determined by the balance between cell proliferation and apoptosis. During the early luteal phase, luteal cells proliferate and differentiate, leading to an increase in progesterone production. However, the corpus luteum undergoes regression in the late luteal phase, leading to a decrease in progesterone production [61,62]. In tumors, apoptosis can occur in two distinct pathways: the intrinsic and extrinsic pathways. The intrinsic pathway is primarily regulated by the balance between pro-apoptotic and anti-apoptotic proteins of the Bcl-2 family. In contrast, anti-apoptotic members, such as Bcl-2 and Bcl-xL, inhibit mitochondrial permeabilization and prevent apoptosis. The extrinsic pathway, also known as the death receptor pathway, is activated by binding specific ligands to death receptors on the cell surface. This triggers the recruitment of adaptor molecules and the activation of caspases, ultimately resulting in cell death [4,9,63,64].
3.1. Apoptosis in Corpus Luteum
In the corpus luteum, apoptosis is a key mechanism for the regression of the structure if pregnancy does not occur [62]. The decline in pro-survival factors, such as insulin-like growth factor 1 (IGF-1) and luteinizing hormone (LH), and the increase in pro-apoptotic factors, including Fas ligand (FasL) and tumor necrosis factor-alpha (TNF-α). FasL and TNF-α trigger apoptotic pathways in the luteal cells [65,66,67]. The apoptosis of luteal cells is primarily regulated by the intrinsic pathway. Pro-apoptotic members of the Bcl-2 family promote mitochondrial outer membrane permeabilization, leading to the release of cytochrome c. Cytochrome c activates caspase, the key effector of apoptosis, resulting in luteal cell death [61,68]. In addition to the intrinsic pathway, the extrinsic pathway can contribute to apoptosis in the corpus luteum. FasL, a ligand that activates the death receptor pathway, is expressed in the regressing corpus luteum. The binding of FasL to its receptor Fas triggers the recruitment of adaptor molecules and the activation of caspases, leading to apoptosis [69].
3.2. Apoptosis in Tumor
In tumors, apoptosis can occur through two distinct pathways: the intrinsic pathway and the extrinsic pathway. The intrinsic pathway is primarily regulated by the balance between pro-apoptotic and anti-apoptotic members, such as Bcl-2 and Bcl-xL, which inhibit mitochondrial permeabilization and prevent apoptosis. The extrinsic pathway, also known as the death receptor pathway, is activated by binding specific ligands to death receptors on the cell surface. This triggers the recruitment of adaptor molecules and the activation of caspases, ultimately resulting in cell death. The tumor necrosis factor-alpha (TNF-α) and Fas ligand (Fas L) are ligands and can activate the extrinsic pathway [70]. In tumor development, apoptosis serves as a critical defense mechanism against cancer. When cells acquire genetic mutations that promote uncontrolled growth, apoptosis acts as a failsafe mechanism to eliminate these aberrant cells [4,64]. However, cancer cells can develop various mechanisms to evade apoptosis, allowing them to survive and propagate. One of the hallmarks of cancer is the dysregulation of the balance between cell proliferation and cell death, favoring cell survival and tumor growth. Cancer cells can upregulate anti-apoptotic proteins or downregulate pro-apoptotic proteins, thereby escaping apoptosis. Moreover, they can disrupt the signaling pathways involved in apoptosis, making themselves resistant to cell death signals [9,70].
3.3. Cellular Interactions
The microenvironments of apoptosis in both the corpus luteum and tumor involve intricate cellular interactions between different cell types. These interactions play critical roles in regulating apoptosis and maintaining tissue integrity. In the corpus luteum, apoptotic cell death occurs during the regression phase if pregnancy does not occur. The luteal cells undergo apoptosis response to changes in hormonal signaling, particularly a decline in pro-survival factors such as insulin-like growth factor 1 (IGF-1) and luteinizing hormone (LH) [3,65]. This apoptotic process is regulated by interactions between luteal cells and immune cells, including macrophages and lymphocytes. Immune cells release cytokines and apoptotic signals that trigger cell death in luteal cells [71]. Similarly, apoptosis can occur in tumors due to intrinsic or extrinsic stimuli. Tumor cells can undergo apoptosis in response to various signals, such as DNA damage, nutrient deprivation, or immune-mediated cell death. The interactions between tumor cells and immune cells, including cytotoxic T cells and natural killer (NK) cells, play a crucial role in regulating apoptosis. Immune cells can release cytotoxic molecules, such as perforin and granzymes, that induce apoptosis in tumor cells [7,72].
3.4. Signaling Molecules
The microenvironments of apoptosis in the corpus luteum and tumor involve the activation of specific signaling pathways and the modulation of various signaling molecules. In the corpus luteum, the regression phase is associated with increased pro-apoptotic factors, such as Fas ligand (FasL) and tumor necrosis factor-alpha (TNF- α). These factors bind to their respective receptors on luteal cells, initiating signaling cascades that lead to apoptotic cell death. The Fas/FasL pathway plays a significant role in regulating apoptosis in the corpus luteum [61,66,67]. Similarly, apoptosis can be induced in tumors through intrinsic or extrinsic pathways. Intrinsic apoptosis is triggered by intracellular signals, such as DNA damage or cellular stress, which activate pro-apoptotic proteins, including Bax and Bak. These proteins promote mitochondrial outer membrane permeabilization (MOMP), leading to the release of cytochrome c and the activation of caspases, the key effectors of apoptosis [73,74]. On the other hand, extrinsic apoptosis is initiated by binding death ligands, such as FasL or TNF-related apoptosis-inducing ligand (TRAIL), to their corresponding death receptors on tumor cells. This binding activates caspase cascades and leads to apoptosis [75,76].
3.5. Extracellular Factors
The microenvironments of apoptosis in the corpus luteum and tumors involve the influence of extracellular factors and the extracellular matrix (ECM) on cell survival and death processes [77]. In the corpus luteum, the ECM undergoes remodeling during the regression phase, leading to changes in its composition and structure. The remodeling process involves the production and activation of various ECM-degrading enzymes, such as matrix metalloproteinases (MMPs). These enzymes facilitate the breakdown of the ECM and contribute to luteal cell apoptosis [78]. Additionally, cytokines and chemokines released by immune cells can modulate [79]. Similarly, the ECM can modulate tumor apoptosis by influencing cell-ECM interactions, cellular signaling, and responses to apoptotic stimuli [80].
3.6. Cytokines
Cytokines are small signaling molecules that regulate cellular response and play important roles in apoptosis. They can influence cell survival, proliferation, and cell death pathways. In the microenvironments of the corpus luteum and tumors, several cytokines are involved in apoptosis. In the corpus luteum, cytokines such as tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ) have been implicated in the regulation of apoptosis. TNF-α can induce apoptosis in luteal cells by activating apoptotic signaling pathways, while IFN-γ can promote apoptosis by sensitizing cells to apoptotic stimuli [71,81]. Similarly, cytokines such as TNF-α, interleukin-1 (IL-1), and interferons can regulate tumor apoptosis. These cytokines can induce apoptosis directly or sensitize tumor cells to apoptotic signals, and they can also modulate the immune response, leading to immune-mediated cell death [82,83].
3.7. Signaling Pathways
3.7.1. Tumor Suppressor Pathways
One of the key regulators of apoptosis in both the corpus luteum and tumors is the tumor suppressor protein p53. P53 acts as a transcription factor and controls the expression of numerous genes involved in cell cycle arrest, DNA repair, and apoptosis. In response to various stresses such as DNA damage, hypoxia, or oncogene activation, p53 is stabilized and activated [62,84]. Activated p53 promotes apoptosis through several mechanisms. It induces the transcription of pro-apoptotic genes, such as Bax and PUMA, which promote mitochondrial outer membrane permeabilization (MOMP) and cytochrome c release. Cytochrome c, along with apoptotic protease-activating factor 1 (Apaf-1) and procaspase-9, forms the apoptosome, leading to the activation of effector caspases and the execution of apoptosis [85,86]. In addition, the retinoblastoma (RB) pathway is another tumor suppressor pathway involved in apoptosis regulation. The RB protein regulates the cell cycle by inhibiting the activity of the E2F transcription factors. Activation of the RB pathway leads to cell cycle arrest and apoptosis induction. Dysregulation of the RB pathway can disrupt the balance between cell proliferation and apoptosis, contributing to tumorigenesis [87].
3.7.2. Mitochondrial Pathway
The mitochondrial pathway is a central mechanism in apoptosis that is shared between the corpus luteum and tumors. The Bcl-2 family of proteins, including anti-apoptotic members (Bcl-2, Bcl-XL) and pro-apoptotic members (Bax, Bak), regulate mitochondrial outer membrane permeabilization (MOMP) and cytochrome c release [73,74,88].
MOMP is a critical event in the intrinsic pathway of apoptosis. It involves the release of apoptogenic factors from the mitochondrial intermembrane space into the cytosol, triggering the activation of downstream apoptotic cascades. Pro-apoptotic Bcl-2 family members, such as Bax and Bak, promote MOMP. Upon receiving apoptotic signals, these proteins undergo conformational changes and translocate to the mitochondrial outer membrane, where they form channels or pores. This leads to the release of cytochrome c and other apoptogenic factors from the mitochondrial intermembrane space. Also, anti-apoptotic Bcl-2 family members, such as Bcl-2 and Bcl-xL, inhibit MOMP and protect cells from apoptosis. They interact with pro-apoptotic proteins, preventing their activation and translocation to the mitochondrial outer membrane. Under normal conditions, anti-apoptotic Bcl-2 family members prevent MOMP by binding to pro-apoptotic members and inhibiting their activity. In both the corpus luteum and tumors, MOMP is regulated by the balance between pro-apoptotic and anti-apoptotic members of the Bcl-2 protein family [89,90].
However, in response to apoptotic stimuli, the balance shifts towards pro-apoptotic proteins, leading to MOMP and cytochrome c release. In both the corpus luteum and tumor, the release of cytochrome c triggers downstream apoptotic signaling events. Upon MOMP, cytochrome c is released from the mitochondrial intermembrane space into the cytosol [89]. Once in the cytosol, cytochrome c interacts with the apoptotic protease-activating factor 1 (Apaf-1) to form the apoptosome. The cytochrome c binds to Apaf-1, promoting the assembly of the apoptosome and the activation of caspase-9. This leads to the activation of effector caspases, such as caspase-3 and caspase-7, resulting in apoptotic cell death [91,92,93].
Moreover, caspases, a family of cysteine proteases, play a central role in the execution of apoptosis. In both the corpus luteum and tumors, caspase activation is a key event downstream of the mitochondrial signaling pathway. Activate caspase-9, formed upon apoptosome assembly, cleaves and activates effector caspases, such as caspase-3, -6, and -7 [92,93,94]. These effector caspases execute the dismantling of the cell by cleaving specific cellular substrates, leading to characteristic apoptotic changes, such as DNA fragmentation, cytoskeletal breakdown, and nuclear condensation [7].
3.7.3. Death Receptor Pathway
The death receptor pathway, the extrinsic pathway, plays a significant role in apoptosis induction in both the corpus luteum and tumors. This pathway is initiated by binding death ligands, such as tumor necrosis factor-alpha (TNF-α) and Fas ligand (FasL), to their corresponding death receptors, TNFR1 and Fas, respectively. Two well-known death receptor systems are the Fas/FasL system and the TNF/TNFR system [4,69]. TNFR1 is a death receptor that binds to TNF-α, a key death ligand. TNF-α is a cytokine involved in various physiological and pathological processes, including apoptosis regulation. The binding of TNF-α to TNF1 initiates the formation of a death-inducing signaling complex (DISC), which leads to apoptosis induction [95]. Fas, also known as CD95 or Apo-1, is another death receptor involved in apoptosis. FasL, the ligand for Fas, is expressed on the surface of cytotoxic T cells and natural killer (NK) cells. The binding of FasL to Fas triggers the assembly of the DISC, leading to apoptosis activation [95,96,97].
The DISC is a critical component of death receptor signaling pathways and is formed upon ligand-receptor interaction. The DISC recruits and activates caspases, the key executioners of apoptosis. Upon ligand binding to death receptors, specialized adaptor proteins, such as Fas-associated death domain protein (FADD) and TNF receptor-associated death domain (TRADD), are recruited. These adaptors, in turn, recruit and activate procaspase-8 or procaspase-10, forming the DISC [63,97].
Caspases are cysteine proteases that play a central role in the execution of apoptosis. In both the corpus luteum and tumors, the activation of caspases is a key event downstream of death receptor signaling pathways. Upon activation, procaspase-8 or procaspase-10 within the DISC undergoes autoproteolytic cleavage, resulting in the formation of active caspase-8 or caspase-10. These activated caspases can directly cleave and activate downstream effector caspases, such as caspase-3, -6, and -7. The activation of effector caspases is a crucial step in the execution phase of apoptosis. Once activated, effector caspases cleave specific cellular substrates, leading to characteristic apoptotic changes [63,94]. Effector caspases cleave various cellular proteins, including nuclear lamins, DNA repair enzymes, and cytoskeletal components. These cleavages result in nuclear fragmentation, DNA degradation, and cytoskeletal breakdown, ultimately leading to cell shrinkage and fragmentation [7,94].
4. Conclusions
Apoptosis and angiogenesis are fundamental processes that play critical roles in developing, maintaining, and regressing various tissues and organs. The corpus luteum is a transient endocrine gland formed in the ovary after ovulation, and tumors are abnormal growths of cells. Both tissues exhibit apoptosis and angiogenesis to fulfill their physiological and pathological functions. Understanding the similarities in the microenvironments that regulate apoptosis and angiogenesis in the corpus luteum and tumors is important with reproductive hormones (Figure 2). Thus, we suggest that these studies can provide valuable insights into the shared mechanisms and interactions within angiogenesis and apoptosis processes, leading to potential therapeutic targets and strategies for various diseases, including reproductive hormone functions in animal and human ovaries.
Author Contributions
T.M. and S.L. conceived, structured, and wrote the manuscript. S.H.L. analyzed data. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The author declares no competing interests.
References
- Robinson, R.S.; Woad, K.J.; Hammond, A.J.; Laird, M.; Hunter, M.G.; Mann, G.E. Angiogenesis and vascular function in the ovary. Reproduction 2009, 138, 869–881. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; Liao, Q.; Xiang, B.; Zhou, M.; Guo, C.; Zeng, Z.; Li, G.; Li, X.; Xiong, W. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Jin, X.; Xiao, L.J.; Zhang, X.S.; Liu, Y.X. Apotosis in ovary. Front. Biosci. 2011, 3, 680–697. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: from pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Okuda, K. Multiple roles of hypoxia in bovine corpus luteum. J. Reprod. Dev. 2020, 66, 307–310. [Google Scholar] [CrossRef]
- Ribatti, D.; Crivellato, E. Immune cells and angiogenesis. J. Cell. Mol. Med. 2009, 13, 2822–2833. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: a review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Vaskivuo, T.E.; Tapanainen, J.S. Apoptosis in the human ovary. Reprod. Biomed. Online 2003, 6, 24–35. [Google Scholar] [CrossRef]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef]
- Gupta, M.K.; Qin, R.Y. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Rizov, M.; Andreeva, P.; Dimova, I. Molecular regulation and role of angiogenesis in reproduction. Taiwan. J. Obstet. Gynecol. 2017, 56, 127–132. [Google Scholar] [CrossRef]
- Gavalas, N.G.; Liontos, M.; Trachana, S.P.; Bagratuni, T.; Arapinis, C.; Liacos, C.; Dimopoulos, M.A.; Bamias, A. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 15885–15909. [Google Scholar] [CrossRef] [PubMed]
- Zalman, Y.; Klipper, E.; Farberov, S.; Mondal, M.; Wee, G.; Folger, J.K.; Smith, G.W.; Meidan, R. Regulation of angiogenesis-related prostaglandin f2alpha-induced genes in the bovine corpus luteum. Biol. Reprod. 2012, 86, 92. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kang, G.; Wang, T.; Huang, H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol. Lett. 2018, 16, 687–702. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Dhanabal, M.; Griffioen, A.W.; Sukhatme, V.P.; Ramakrishnan, S. Synergy between angiostatin and endostatin: inhibition of ovarian cancer growth. Cancer Res. 2000, 60, 2190–2196. [Google Scholar] [PubMed]
- Sugino, N.; Matsuoka, A.; Taniguchi, K.; Tamura, H. Angiogenesis in the human corpus luteum. Reprod. Med. Biol. 2008, 7, 91–103. [Google Scholar] [CrossRef]
- Devesa, J.; Caicedo, D. The Role of Growth Hormone on Ovarian Functioning and Ovarian Angiogenesis. Front. Endocrinol. 2019, 10, 450. [Google Scholar] [CrossRef]
- Schams, D.; Berisha, B. Angiogenic Factors (VEGF, FGF and IGF) in the Bovine Corpus Luteum. J. Reprod. Dev. 2002, 48, 233–242. [Google Scholar] [CrossRef]
- Takenaga, K. Angiogenic signaling aberrantly induced by tumor hypoxia. Front. Biosci. 2011, 16, 31–48. [Google Scholar] [CrossRef]
- Samples, J.; Willis, M.; Klauber-Demore, N. Targeting angiogenesis and the tumor microenvironment. Surg. Oncol. Clin. N. Am. 2013, 22, 629–639. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Betsholtz, C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003, 314, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Murdoch, C. Macrophage responses to hypoxia: implications for tumor progression and anti-cancer therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ji, Y.R.; Lee, Y.M. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch. Pharm. Res. 2022, 45, 401–416. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Shirasuna, K.; Shimizu, T.; Matsui, M. Impact of angiogenic and innate immune systems on the corpus luteum function during its formation and maintenance in ruminants. Reprod. Biol. 2013, 13, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Yee, K.O.; Lawler, J.; Khosravi-Far, R. Regulation of tumor angiogenesis by thrombospondin-1. Biochim. Biophys. Acta 2006, 1765, 178–188. [Google Scholar] [CrossRef]
- Plendl, J. Angiogenesis and vascular regression in the ovary. Anat Histol Embryol 2000, 29, 257–266. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, dev151019. [Google Scholar] [CrossRef]
- Senger, D.R.; Davis, G.E. Angiogenesis. Cold Spring Harbor Perspect. Biol. 2011, 3, a005090. [Google Scholar] [CrossRef]
- Lepucki, A.; Orlińska, K.; Mielczarek-Palacz, A.; Kabut, J.; Olczyk, P.; Komosińska-Vassev, K. The Role of Extracellular Matrix Proteins in Breast Cancer. J. Clin. Med. 2022, 11, 1250. [Google Scholar] [CrossRef]
- Conway, E.M.; Collen, D.; Carmeliet, P. Molecular mechanisms of blood vessel growth. Cardiovasc. Res. 2001, 49, 507–521. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Matsuo, Y.; Takeyama, H.; Guha, S. Cytokine network: new targeted therapy for pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2416–2419. [Google Scholar] [CrossRef]
- Li, A.; Varney, M.L.; Valasek, J.; Godfrey, M.; Dave, B.J.; Singh, R.K. Autocrine role of interleukin-8 in induction of endothelial cell proliferation, survival, migration and MMP-2 production and angiogenesis. Angiogenesis 2005, 8, 63–71. [Google Scholar] [CrossRef]
- Jiemtaweeboon, S.; Shirasuna, K.; Nitta, A.; Kobayashi, A.; Schuberth, H.J.; Shimizu, T.; Miyamoto, A. Evidence that polymorphonuclear neutrophils infiltrate into the developing corpus luteum and promote angiogenesis with interleukin-8 in the cow. Reprod. Biol. Endocrinol. 2011, 9, 79. [Google Scholar] [CrossRef]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef] [PubMed]
- Galvão, A.M.; Ferreira-Dias, G.; Skarzynski, D.J. Cytokines and angiogenesis in the corpus luteum. Mediat. Inflamm. 2013, 2013, 420186. [Google Scholar] [CrossRef] [PubMed]
- Ono, M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D.I.; Zachary, I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef]
- Geva, E.; Jaffe, R.B. Role of vascular endothelial growth factor in ovarian physiology and pathology. Fertil. Steril. 2000, 74, 429–438. [Google Scholar] [CrossRef]
- Fraser, H.M.; Wulff, C. Angiogenesis in the corpus luteum. Reprod. Biol. Endocrinol. 2003, 1, 88. [Google Scholar] [CrossRef]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumor angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signaling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Molec. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010, 21, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S.; Rueda, B.R.; Spanel-Borowski, K. Microvascular endothelial cells of the corpus luteum. Reprod. Biol. Endocrinol. 2003, 1, 89. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signaling: from development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Yang, X.; Liaw, L.; Prudovsky, I.; Brooks, P.C.; Vary, C.; Oxburgh, L.; Friesel, R. Fibroblast growth factor signaling in the vasculature. Curr. Atheroscleros. Rep. 2015, 17, 509. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, J.B.; Essaghir, A. PDGF receptor signaling networks in normal and cancer cells. Cytokine Growth Factor Rev. 2014, 25, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Tang, X.Y.; Qu, Z.Y.; Sun, Z.W.; Ji, C.F.; Li, Y.J.; Guo, S.D. Targeting the PDGF/PDGFR signaling pathway for cancer therapy: A review. Int. J. Biol. Macromol. 2022, 202, 539–557. [Google Scholar] [CrossRef]
- Kazlauskas, A. PDGFs and their receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Niba, E.T.; Nagaya, H.; Kanno, T.; Tsuchiya, A.; Gotoh, A.; Tabata, C.; Kuribayashi, K.; Nakano, T.; Nishizaki, T. Crosstalk between PI3 kinase/PDK1/Akt/Rac1 and Ras/Raf/MEK/ERK pathways downstream PDGF receptor. Cell Physiol Biochem 2013, 31, 905–913. [Google Scholar] [CrossRef]
- Razmara, M.; Heldin, C.H.; Lennartsson, J. Platelet-derived growth factor-induced Akt phosphorylation requires mTOR/Rictor and phospholipase C-γ1, whereas S6 phosphorylation depends on mTOR/Raptor and phospholipase D. Cell Commun. Signal. 2013, 11, 3. [Google Scholar] [CrossRef]
- Hojo, T.; Skarzynski, D.J.; Okuda, K. Apoptosis, autophagic cell death, and necroptosis: different types of programmed cell death in bovine corpus luteum regression. J. Reprod. Dev. 2022, 68, 355–360. [Google Scholar] [CrossRef]
- Niswender, G.D.; Juengel, J.L.; Silva, P.J.; Rollyson, M.K.; McIntush, E.W. Mechanisms controlling the function and life span of the corpus luteum. Physiol. Rev. 2000, 80, 1–29. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging-US 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Balogh, O.; Müller, L.; Boos, A.; Kowalewski, M.P.; Reichler, I.M. Expression of insulin-like growth factor 1 and its receptor in preovulatory follicles and in the corpus luteum in the bitch. Gen. Comp. Endocrinol. 2018, 269, 68–74. [Google Scholar] [CrossRef]
- Galvao, A.M.; Ramilo, D.W.; Skarzynski, D.J.; Lukasik, K.; Tramontano, A.; Mollo, A.; Mateus, L.M.; Ferreira-Dias, G.M. Is FAS/Fas ligand system involved in equine corpus luteum functional regression? Biol. Reprod. 2010, 83, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Sakumoto, R.; Berisha, B.; Kawate, N.; Schams, D.; Okuda, K. Tumor necrosis factor-alpha and its receptor in bovine corpus luteum throughout the estrous cycle. Biol. Reprod. 2000, 62, 192–199. [Google Scholar] [CrossRef]
- Antonsson, B. Bax and other pro-apoptotic Bcl-2 family “killer-proteins” and their victim the mitochondrion. Cell Tissue Res. 2001, 306, 347–361. [Google Scholar] [CrossRef]
- Okuda, K.; Sakumoto, R. Multiple roles of TNF superfamily members in corpus luteum function. Reprod. Biol. Endocrinol. 2003, 1, 95. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Pate, J.L.; Landis Keyes, P. Immune cells in the corpus luteum: friends or foes? Reproduction 2001, 122, 665–676. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R. Mechanisms of tumor escape: role of tumor microenvironment in inducing apoptosis of cytolytic effector cells. Arch. Immunol. Ther. Exp. 2006, 54, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yang, J.; Jones, D.P. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim. Biophys. Acta. 1998, 1366, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway signaling. J. Thorac. Oncol. 2007, 2, 461–465. [Google Scholar] [CrossRef]
- Waring, P.; Müllbacher, A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell Biol. 1999, 77, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Lee, J.H.; Yoon, J.T. Expression of matrix metalloproteinases to induce the expression of genes associated with apoptosis during corpus luteum development in bovine. PeerJ 2019, 7, e6344. [Google Scholar] [CrossRef]
- Boyd, D.F.; Thomas, P.G. Towards integrating extracellular matrix and immunological pathways. Cytokine 2017, 98, 79–86. [Google Scholar] [CrossRef]
- Folgueras, A.R.; Pendás, A.M.; Sánchez, L.M.; López-Otín, C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int. J. Dev. Biol. 2004, 48, 411–424. [Google Scholar] [CrossRef]
- Petroff, M.G.; Petroff, B.K.; Pate, J.L. Mechanisms of cytokine-induced death of cultured bovine luteal cells. Reproduction 2001, 121, 753–760. [Google Scholar] [CrossRef]
- Shen, J.; Xiao, Z.; Zhao, Q.; Li, M.; Wu, X.; Zhang, L.; Hu, W.; Cho, C.H. Anti-cancer therapy with TNFα and IFNγ: A comprehensive review. Cell Prolif. 2018, 51, e12441. [Google Scholar] [CrossRef] [PubMed]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L. The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. [Google Scholar] [CrossRef]
- Meulmeester, E.; Jochemsen, A.G. p53: a guide to apoptosis. Curr. Cancer Drug Targets 2008, 8, 87–97. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Nevins, J.R. The Rb/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Tsujimoto, Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells 1998, 3, 697–707. [Google Scholar] [CrossRef]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Kornbluth, S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev. Cell 2006, 10, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Apoptosome: the cellular engine for the activation of caspase-9. Structure 2002, 10, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harbor Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Ukrainskaya, V.M.; Stepanov, A.V.; Glagoleva, I.S.; Knorre, V.D.; Belogurov, A.A.J.; Gabibov, A.G. Death Receptors: New Opportunities in Cancer Therapy. Acta Naturae 2017, 9, 55–63. [Google Scholar] [CrossRef]
- Kojima, Y.; Kawasaki-Koyanagi, A.; Sueyoshi, N.; Kanai, A.; Yagita, H.; Okumura, K. Localization of Fas ligand in cytoplasmic granules of CD8+ cytotoxic T lymphocytes and natural killer cells: participation of Fas ligand in granule exocytosis model of cytotoxicity. Biochem. Biophys. Res. Commun. 2002, 296, 328–336. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Krammer, P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012, 19, 36–41. [Google Scholar] [CrossRef]
Figure 1.
Angiogenesis- and apoptosis-related factors in corpus luteum and tumor. In corpus luteum and tumor, hormones, angiogenic growth, extracellular, cytokines, apoptotic, and cytotoxic factors contribute to angiogenesis and apoptosis. Black letters, tumor- and CL-regulated; Red letters, CL-regulated; Blue letters, tumor-regulated; CL, corpus luteum.
Figure 1.
Angiogenesis- and apoptosis-related factors in corpus luteum and tumor. In corpus luteum and tumor, hormones, angiogenic growth, extracellular, cytokines, apoptotic, and cytotoxic factors contribute to angiogenesis and apoptosis. Black letters, tumor- and CL-regulated; Red letters, CL-regulated; Blue letters, tumor-regulated; CL, corpus luteum.
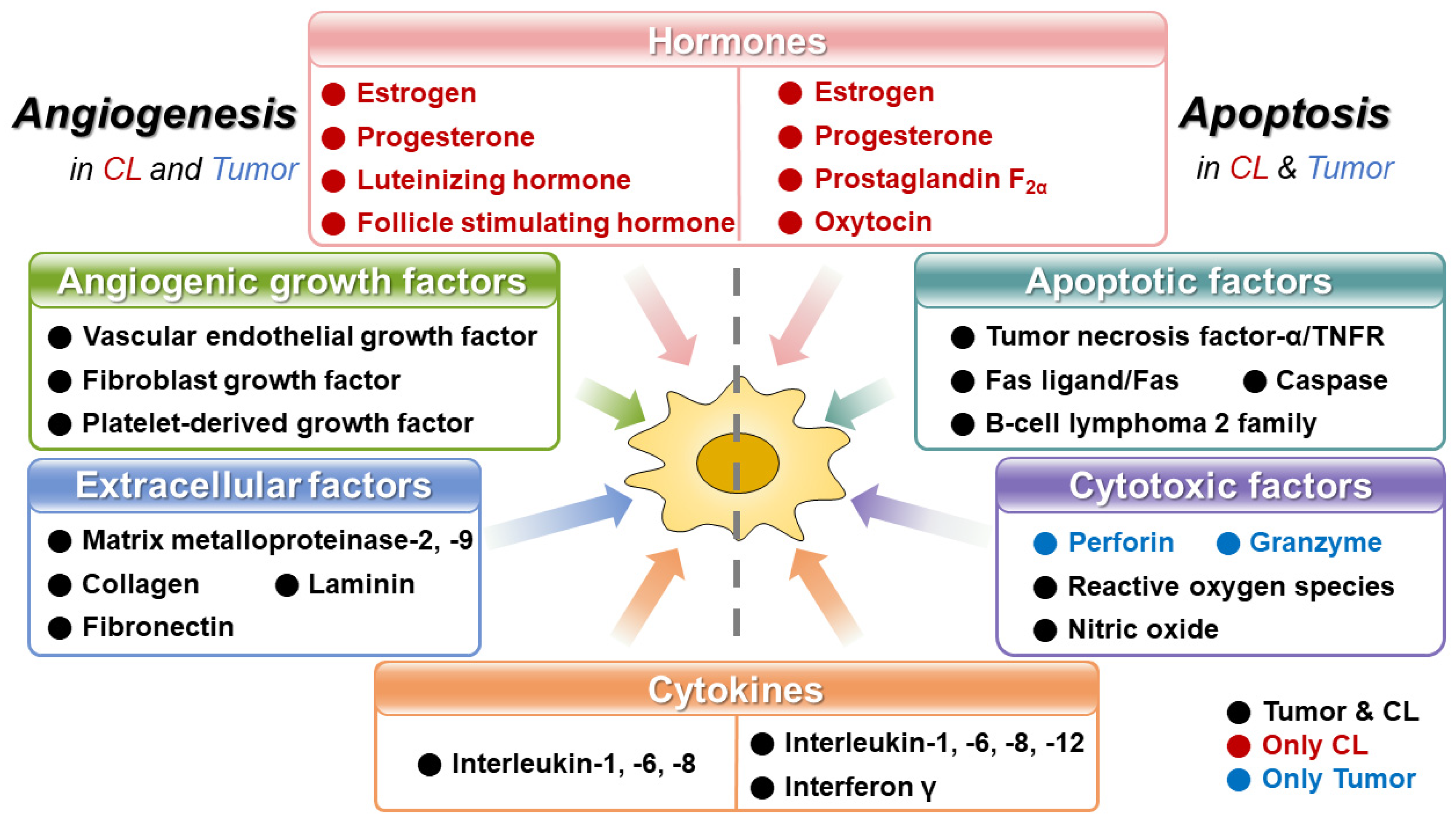
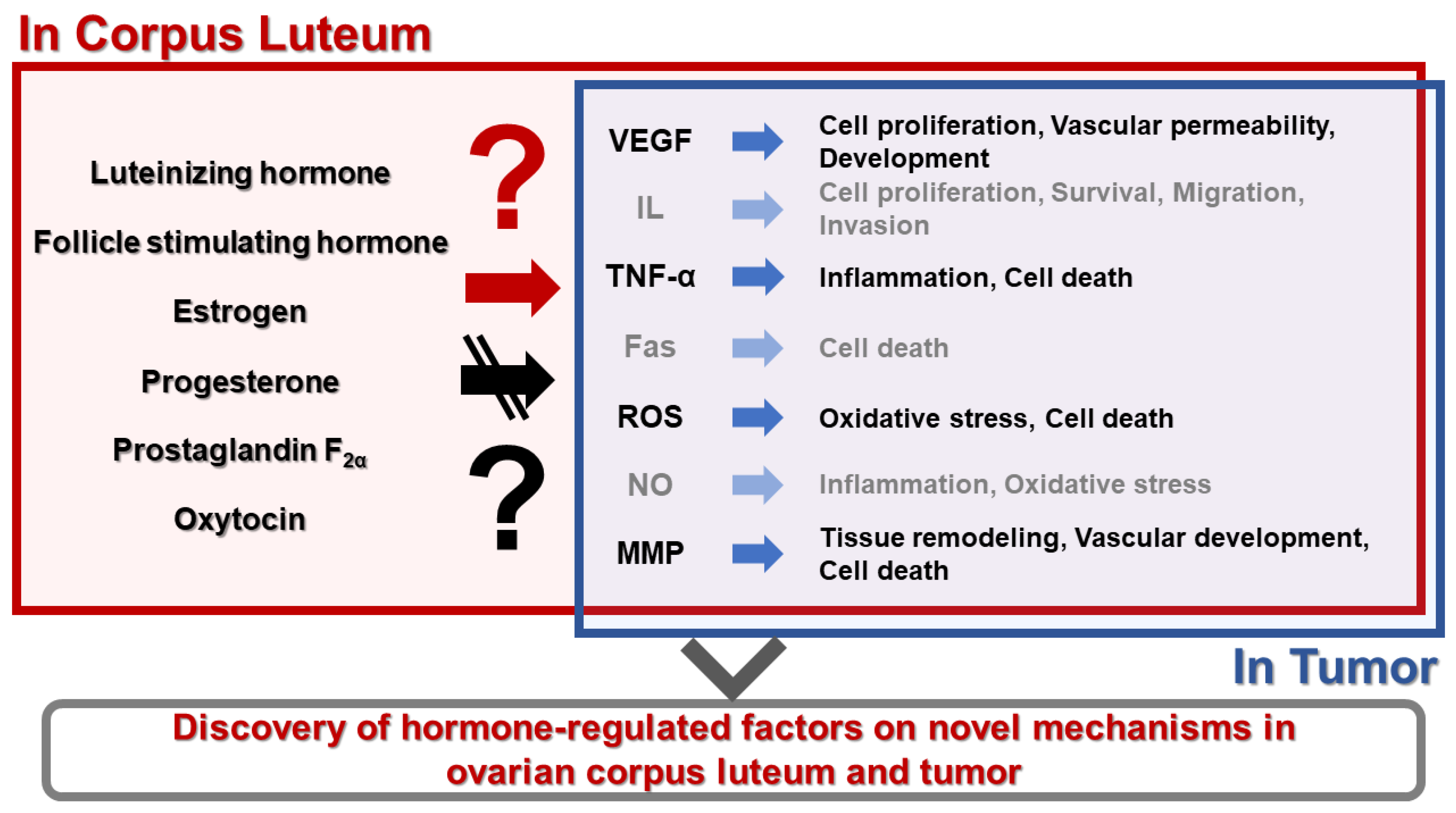
Figure 2.
Diagram of the novel model on the hormone-regulated factors in the physiology function of the ovarian corpus luteum and tumor. Hormones play a critical role in the formation and regression of the corpus luteum by regulating several factors. This model shows a new research area that applies mechanisms of hormones in tumor physiology. VEGF, vascular endothelial growth factor; IL, interleukin; TNF-α, tumor necrosis factor-α; ROS, reactive oxygen species; NO, nitric oxide; MMP, matrix metalloproteinase.
Figure 2.
Diagram of the novel model on the hormone-regulated factors in the physiology function of the ovarian corpus luteum and tumor. Hormones play a critical role in the formation and regression of the corpus luteum by regulating several factors. This model shows a new research area that applies mechanisms of hormones in tumor physiology. VEGF, vascular endothelial growth factor; IL, interleukin; TNF-α, tumor necrosis factor-α; ROS, reactive oxygen species; NO, nitric oxide; MMP, matrix metalloproteinase.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



