
Submitted:
10 March 2024
Posted:
11 March 2024
You are already at the latest version
Abstract
Regulatory T cells are key immune cells to initiate tolerable immune responses. TGFβ is the key cytokine of regulatory T cells. TGFβ is also the master cytokine to stimulate tissue fibrosis. Systemic sclerosis is an autoimmune disease related to overt organ fibrosis. Thus, systemic sclerosis is a disease related to regulatory T cell over-representation. Besides, many end stage organ failures including heart failure, liver cirrhosis, uremia, and pulmonary fibrosis also related to tissue fibrosis. Thus, TGFβ producing regulatory T cells are also involved in the pathogenesis of these end stage organ failure. Once we know the significance of TGFβ and it induced tolerable immune reactions in the pathophysiology of end organ failure, we can develop better prevention or therapeutic strategies to combat these detrimental disorders.
Keywords:
Treg
; TGFβ
; Scleroderma
; heart failure
; liver cirrhosis
; uremia
; pulmonary fibrosis
Introduction
Host immunological pathways can be categorized into IgG dominant eradicable immune responses and IgA dominant tolerable immune responses[1-4]. Regulatory CD4CD25 T cells (Treg) are key initiator cells to drive host tolerable immune responses. Treg cells can produce TGFβ to facilitate the tolerable immune reactions like TH17, TH9, TH1-like, and TH3 immune reactions. For examples: TH17 immune reaction is triggered by TGFβ plus interleukin-6. TH9 immune reaction is triggered by TGFβ plus interleukin-4. There is a misleading that Treg cells cause profound immunosuppression. Actually, the physiological role of profound immunosuppression is caused by corticosteroid. The physiological role of TGFβ is to initiate tolerable IgA dominant immune reactions to use milder control strategies to deal with chronic or extensive pathogen infections. IgA is a weak antibody to avoid profound severe host immune reactions to kill cells, tissues, or organs in extensive pathogen infections. Another function of Treg cells is to mediate tissue repair. TGFβ is the master regulator of tissue fibrosis to use collagens and other extracellular matrix to repair wounds or lost tissues or cells. Thus, there is a link between tissue fibrosis and Treg dominant tolerable host immune reactions. Tissue fibrosis is also a hallmark of scleroderma and end organ failures. Here, we will use literature review to point out the Treg dominant tolerable host immune reactions and their link to scleroderma and end organ failures.
The Intricate Tapestry of Host Immunological Pathways
The human immune system is a remarkably complex and intricate tapestry, woven by a myriad of interconnected pathways designed to safeguard the body against a diverse array of potential threats. At the heart of this immunological labyrinth lies a fundamental dichotomy: the eradicable immune responses, dominated by the IgG antibody isotype, and the tolerable immune responses, governed by the IgA antibody isotype[1].
The Eradicable Immune Responses: A Formidable Force
The eradicable immunological pathways represent the body's formidable defense mechanisms, meticulously orchestrated to eliminate pathogenic invaders. These pathways are initiated by a specialized subset of T cells known as follicular helper T cells (Tfh), characterized by their expression of the CXCR5 chemokine receptor and their secretion of interleukin-21 (IL-21). The transcription factors BCL6 and STAT5B play pivotal roles in mediating these immune reactions.
The primary function of Tfh cells is to promote the production of antibodies by B cells within the germinal centers of lymphoid tissues. Crucially, they facilitate the class switch from IgM to IgG antibodies, a process mediated by the potent cytokine IL-21. This class switch is a critical step in the development of eradicable immune responses, as IgG antibodies are particularly adept at neutralizing and eliminating pathogens.
Within the eradicable immune responses, four distinct branches have evolved to combat four specific categories of pathogens:
- The TH1 Immunological Pathway: Combating Intracellular Invaders
The TH1 immunological pathway is the body's defensive line against intracellular microorganisms, such as intracellular bacteria, fungi, and protozoa. Key players in this pathway include type 2 myeloid dendritic cells (mDC2), type 1 innate lymphoid cells (ILC1), type 1 macrophages (M1), IFN-γ-secreting CD4+ T cells, CD8+ T cells (Tc1), type 1 invariant natural killer T cells (iNKT1), and IgG3-producing B cells.
Driven by the cytokine interleukin-12 (IL-12), the TH1 immune response is governed by the transcription factors STAT4 and STAT1. The effector cytokine IFN-γ activates M1 macrophages via inducible nitric oxide synthase (iNOS), leading to the generation of free radicals that cause lipid membrane peroxidation, ultimately killing the ingested microorganisms. This pathway is associated with type IV delayed-type hypersensitivity reactions.
- 2.
- The TH2 Immunological Pathway: Combating Parasitic Foes
The TH2 immunological pathway is the body's defense against parasitic invaders, further subdivided into two distinct subtypes: TH2a and TH2b. TH2a immunity targets endoparasites, such as helminths, while TH2b immunity targets ectoparasites, like insects.
In the TH2a immune response, Langerhans cells serve as the antigen-presenting cells, while type 2 interleukin-25-inducing natural innate lymphoid cells (nILCs2) are the innate lymphoid cells involved. The key cytokines are interleukin-4 (IL-4) and interleukin-5 (IL-5), with STAT6 and STAT1 as the key transcription factors. The major effector cells include inflammatory eosinophils (iEOS), mast cells-tryptase (MCt), IL-4/IL-5-producing CD4+ T cells, iNKT2 cells, and IgG4-producing B cells.
In the TH2b immune response, Langerhans cells remain the antigen-presenting cells, while type 2 interleukin-33-inducing inflammatory innate lymphoid cells (iILCs2) are the innate lymphoid cells involved. The key cytokines are IL-4 and interleukin-13 (IL-13), with STAT6 and STAT3 as the key transcription factors. The major effector cells include basophils, mast cells-tryptase/chymase (MCtc), IL-4/IL-13-producing CD4+ T cells, iNKT2 cells, and IgE-producing B cells.
The TH2 immunological pathway is associated with type I allergic hypersensitivity reactions, with TH2a immunity related to IgG4-dominant allergy and TH2b immunity related to IgE-dominant allergy.
- 3.
- The TH22 Immunological Pathway: Combating Extracellular Threats
The TH22 immunological pathway represents the body's immunity against extracellular microorganisms, such as extracellular bacteria, fungi, and protozoa. In this pathway, type 1 myeloid dendritic cells (mDC1) serve as the antigen-presenting cells, while type 3 NKp46+ innate lymphoid cells (NCR+ ILC3) are the innate lymphoid cells involved.
The effector immune cells in this pathway include neutrophils (N1), IL-22-secreting CD4+ T cells, and IgG2-producing B cells. Driven by the cytokines interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), the effector cytokine is IL-22, governed by the transcription factors STAT3 and STAT4.
Neutrophil activation, including phagocytosis and NETosis (neutrophil extracellular trap formation), can effectively destroy extracellular microorganisms. Additionally, free radical generation during neutrophil phagocytosis can cause membrane lipid peroxidation, leading to the killing of these pathogens. This pathway is associated with type III immune complex-mediated hypersensitivity reactions.
- 4.
- The THαβ Immunological Pathway: Combating Infectious Particles
The THαβ immunological pathway represents the body's defense against infectious particles, such as viruses and prions. Plasmacytoid dendritic cells serve as the antigen-presenting cells, while interleukin-10-producing innate lymphoid cells (ILC10) are the innate lymphoid cells involved.
The effector immune cells in this pathway include natural killer cells (NK1), IL-10-producing CD4+ T cells, CD8+ T cells (Tc2), and IgG1-producing B cells. Driven by type I interferons and IL-10, the major effector cytokine is IL-10, governed by the transcription factors STAT1, STAT1, and STAT3.
Natural killer cell-mediated antibody-dependent cellular cytotoxicity (ADCC) with IgG1 antibodies is the effector function of the THαβ immunity, leading to the apoptosis of virus- or prion-infected cells. During apoptosis, DNA fragmentation destroys viral DNA or RNA, while protein digestion via caspases degrades prion pathogen proteins. This pathway is associated with type II antibody-dependent cellular cytotoxic hypersensitivity reactions.
The Tolerable Immune Responses: A Delicate Balance
While the eradicable immune responses represent the body's formidable defenses, there are instances where complete eradication of pathogens may prove challenging or potentially damaging to the host. In these scenarios, the body initiates tolerable immunological pathways, governed by regulatory CD4+CD25+ T cells (Treg).
These FOXP3+ Treg cells produce transforming growth factor-beta (TGF-β), which activates STAT5 and STAT5, thereby triggering tolerable immunity. Notably, TGF-β also induces B cell antibody isotype class switching to IgA, the hallmark of tolerable immune responses. Other immune cells related to Treg cells include DCreg, Breg, and ILCreg.
In situations where pathogen infections are severe or diverse, making complete eradication challenging due to the potential for severe organ damage or failure, the host initiates these tolerable immunological pathways as a coping mechanism. These pathways are further categorized into four distinct branches:
- The TH1-like Immunological Pathway: Tolerating Intracellular Invaders
The TH1-like immunological pathway represents the host's tolerable immune response against intracellular microorganisms, such as intracellular bacteria, fungi, and protozoa. The effector cells include macrophages (M2), IFN-γ/TGF-β-secreting CD4+ T cells, CD8+ T cells, iNKT1 cells, and IgA1-producing B cells.
The antigen-presenting cells are type 2 myeloid dendritic cells, while the innate lymphoid cells involved are type 1 non-cytotoxic innate lymphoid cells (NCR- ILCs1). The driving cytokines are IL-12 and TGF-β. This pathway is associated with type IV delayed-type hypersensitivity reactions.
- 2.
- The TH9 Immunological Pathway: Tolerating Parasitic Foes
The TH9 immunological pathway represents the host's tolerable immune response against parasites, including both ectoparasites and endoparasites. The effector cells include regulatory eosinophils (rEOS), basophils, mast cells (MMC9), IL-9-secreting CD4+ T cells, iNKT2 cells, and IgA2-producing B cells.
Langerhans cells serve as the antigen-presenting cells, while thymic stromal lymphopoietin (TSLP)-inducing type 2 innate lymphoid cells are the innate lymphoid cells involved. The driving cytokines are IL-4 and TGF-β. This pathway is associated with type I allergic hypersensitivity reactions.
- 3.
- The TH17 Immunological Pathway: Tolerating Extracellular Threats
The TH17 immunological pathway represents the host's tolerable immune response against extracellular microorganisms, such as extracellular bacteria, fungi, and protozoa. The effector cells include neutrophils (N2), IL-17-secreting CD4+ T cells, iNKT17 cells, and IgA2-producing B cells.
Type 1 myeloid dendritic cells serve as the antigen-presenting cells, while type 3 non-cytotoxic innate lymphoid cells (NCR- ILCs3) are the innate lymphoid cells involved. The driving cytokines are IL-6 and TGF-β. This pathway is associated with type III immune complex-mediated hypersensitivity reactions.
- 4.
- The TH3 Immunological Pathway: Tolerating Infectious Particles
The TH3 immunological pathway represents the host's immunity against infectious particles, such as viruses and prions. Plasmacytoid dendritic cells serve as the antigen-presenting cells, while IL-10-producing innate lymphoid cells (ILC10) are the innate lymphoid cells involved.
The effector immune cells include natural killer cells (NK2), IL-10/TGF-β-producing CD4+ T cells, CD8+ T cells, and IgA1-producing B cells. The driving cytokines are TGF-β and IL-10, with IL-10 and TGF-β being the major effector cytokines. The key transcription factors are STAT1, STAT1, STAT3, and STAT5α/β. This pathway is associated with type II antibody-dependent cellular cytotoxic hypersensitivity reactions.
The Clonal Anergy Mechanism: Distinguishing Self from Non-Self
At the heart of the immune system's intricate tapestry lies a fundamental mechanism that allows it to distinguish between foreign antigens and self-antigens: the clonal anergy mechanism. This process ensures that immune cells do not generate an immune response against the body's own tissues, thereby preventing autoimmune disorders.
Each T cell or B cell is capable of recognizing only one specific antigen, a phenomenon known as the clonal mechanism. If a clonal T cell or B cell recognizes a self-antigen, it will generate no immune response, a state referred to as clonal anergy.
The clonal anergy mechanism is believed to be mediated by gamma delta T cells or IgD B cells. Gamma delta T cells develop in the thymus earlier than the development of alpha beta T cells. Thus, if a T cell clone recognizes a self-antigen, it will differentiate into gamma delta T cells first. Consequently, the later development of alpha beta T cells will not recognize self-antigens, and they will only respond to foreign antigens. When gamma delta T cells recognize self-antigens, they induce clonal anergy.
A similar mechanism applies to B lymphocytes. Mature B cells co-express IgD and IgM antibodies on their surface. When a self-antigen is recognized by the IgD antibody on the B cell surface, it triggers clonal anergy with no immune response. However, when a foreign antigen is recognized by the IgM antibody on the B cell surface, it prompts an immune response against the foreign pathogen. Subsequently, the IgM-bearing B cell can undergo antibody isotype class switching to IgG, IgE, or IgA antibodies.
This exquisitely orchestrated clonal anergy mechanism serves as a safeguard, ensuring that the immune system's formidable defenses are directed solely against foreign threats while preserving tolerance to the body's own tissues.
In conclusion, the host immunological pathways represent a remarkably complex and intricate network, meticulously woven to protect the body from a diverse array of potential threats. The eradicable immune responses, dominated by IgG antibodies, serve as the body's formidable defenses, poised to eliminate pathogenic invaders. In contrast, the tolerable immune responses, governed by IgA antibodies, provide a delicate balance, managing chronic or severe infections while minimizing collateral damage to the host. This intricate tapestry is further enriched by the clonal anergy mechanism, which ensures that the immune system's potent defenses are directed solely against foreign threats, preserving tolerance to the body's own tissues. This comprehensive immunological framework exemplifies the remarkable adaptability and sophistication of the human immune system, a marvel of evolutionary design.
The Enigmatic Role of Regulatory T Cells in Organ Fibrosis and Failure
Within the intricate tapestry of the human immune system, a delicate balance exists between eradicable and tolerable immune responses, each serving a distinct purpose in safeguarding the body. Eradicable immune responses, dominated by IgG antibodies, represent the body's formidable defenses, poised to eliminate pathogenic invaders. In contrast, tolerable immune responses, governed by IgA antibodies, provide a delicate equilibrium, managing chronic or severe infections while minimizing collateral damage to the host.
At the heart of these tolerable immune responses lies a specialized subset of cells, the regulatory T cells (Tregs), orchestrating a symphony of immunological processes that can have profound implications for organ health and function.
Regulatory T Cells in Systemic Sclerosis
Systemic sclerosis, or scleroderma, is a debilitating autoimmune disorder characterized by widespread fibrosis of the affected tissues. This condition can manifest in two major subgroups: diffuse cutaneous scleroderma and limited cutaneous scleroderma, distinguished by the degree of trunk involvement. The limited cutaneous form often presents with a subset called CREST syndrome, encompassing calcinosis cutis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia.
Regardless of the subtype, scleroderma is defined by the replacement of normal tissue architecture with rigid, acellular connective tissues. This fibrosis can affect multiple organs, including the skin, lungs, gastrointestinal tract, kidneys, and heart, leading to a myriad of functional impairments and potential life-threatening complications.
Intriguingly, regulatory T cells (Tregs) play a pivotal role in the pathogenesis of systemic sclerosis. These cells, characterized by the expression of the FOXP3 transcription factor, produce transforming growth factor-beta (TGF-β), a potent mediator of tissue fibrosis[5]. TGF-β is often referred to as the "master regulator" of fibrosis, capable of stimulating fibroblast proliferation and promoting epithelial-mesenchymal transition, a process implicated in the development of scleroderma[6].
Evidence from mouse models and clinical trials further underscores the significance of TGF-β in scleroderma[7]. Anti-TGF-β treatments have been shown to prevent skin and lung fibrosis in animal studies, while clinical trials have demonstrated the potential of these therapies to alleviate the symptoms of systemic sclerosis[7].
Moreover, the polymorphism of the FOXP3 gene, the major transcription factor of Treg cells, has been associated with the incidence of systemic sclerosis, further highlighting the intricate link between these cells and the disease pathogenesis[8].
The presence of both IgG and IgA autoantibodies in scleroderma patients provides additional insight into the role of Tregs. IgG antibodies are associated with eradicable immune responses, while IgA antibodies are linked to Treg-induced tolerable immune reactions. The presence of both antibody isotypes suggests that both eradicable and tolerable immune responses may contribute to the pathogenesis of systemic sclerosis.
Furthermore, the levels of interleukin-2 and interleukin-35, cytokines known to promote Treg cell activity, are elevated in scleroderma patients, further supporting the involvement of these cells in the disease process[9].
Regulatory T Cells and the Heart Failure
Heart failure, a debilitating condition characterized by the heart's inability to adequately pump blood, can have devastating consequences on overall health and quality of life. Symptoms such as shortness of breath, excessive fatigue, and lower leg edema are common, while complications like pulmonary edema, respiratory failure, and cardiac arrhythmias can be life-threatening.
Chronic inflammation and subsequent cardiac fibrosis are major contributors to the development of heart failure. Remarkably, regulatory T cells and the associated TGF-β signaling pathway play a crucial role in this fibrotic process.
TGF-β signaling has been shown to stimulate cardiac fibroblast proliferation and promote heart fibrosis. In animal models, elevated TGF-β levels have been linked to cardiac remodeling and the progression of heart failure[10]. Moreover, the polymorphism of the TGF-β gene has been associated with end-stage heart failure, further emphasizing its significance in the disease pathogenesis[11].
The transcription factor SMAD7, known to inhibit TGF-β signaling, has been demonstrated to prevent post-infarction heart failure, highlighting the potential therapeutic implications of targeting this pathway[12].
Additionally, microRNAs and other non-coding RNAs that interact with TGF-β have been shown to influence the cardiac fibrosis process in heart failure, further underscoring the intricate interplay between these regulatory mechanisms.
Interestingly, tolerable immune responses initiated by regulatory T cells, such as TH17 and TH9 immunity, have also been implicated in the pathogenesis of heart failure. The central cytokines of these pathways, interleukin-17 and interleukin-9, respectively, have been linked to cardiac fibrosis, remodeling, and the progression of heart failure[13,14].
Regulatory T Cells and Liver Cirrhosis
Liver cirrhosis, the end-stage of liver failure, is characterized by extensive liver fibrosis and the subsequent development of complications such as jaundice, esophageal varices, ascites, and splenomegaly. The underlying causes of liver cirrhosis are diverse, ranging from alcoholic liver disease and non-alcoholic steatohepatitis to chronic viral hepatitis and drug abuse.
Chronic inflammation of the liver is a common pathway leading to cirrhosis, and TGF-β plays a pivotal role in the fibrotic process. Elevated levels of TGF-β have been observed in patients with liver cirrhosis, and this cytokine has also been implicated in the promotion of hepatocellular carcinoma, a malignancy often accompanied by cirrhosis[15].
Regulatory T cells and the associated chronic tolerable immune responses have been identified as key players in the pathophysiology of liver cirrhosis. Treg cell expansion has been observed in acute decompensation of liver cirrhosis, as well as in cirrhosis induced by hepatitis B virus (HBV) infection and Schistosoma-related liver fibrosis[15,16]. Additionally, Tregs have been implicated in primary biliary cirrhosis, another liver cirrhosis-related disorder[17].
Furthermore, IgA antibodies, which are associated with Treg-induced tolerable immune responses, are highly elevated in liver cirrhosis. The characteristic β-γ bridging pattern observed in serum electrophoresis, due to elevated serum IgA antibodies, is a hallmark of liver cirrhosis. IgA vasculitis and IgA nephropathy have also been correlated with the development of liver cirrhosis, further underscoring the link between IgA and this debilitating condition[18].
Notably, TH17 immune responses, which are initiated by regulatory T cells, have been reported to be upregulated in liver cirrhosis, further emphasizing the intricate interplay between Tregs and organ fibrosis[16].
Regulatory T Cells and Uremia
Uremia, or chronic renal failure, represents the end-stage of kidney dysfunction, characterized by the impaired ability of the kidneys to remove metabolic waste products from the body. Without intervention through dialysis or kidney transplantation, uremia can progress to life-threatening complications such as coma and death.
Underlying conditions that can lead to chronic renal failure include diabetes, hypertension, glomerulonephritis, interstitial nephritis, polycystic kidney disease, and recurrent urinary tract infections. Chronic inflammation is a common pathway contributing to the development of uremia.
Interestingly, IgA nephropathy, a condition characterized by the presence of IgA antibodies, frequently leads to chronic renal failure. IgA antibodies are associated with Treg-induced tolerable immune reactions, suggesting a potential link between Tregs and the pathogenesis of uremia[19,20].
TGF-β, produced by regulatory T cells, plays a crucial role in the progression of renal fibrosis, a hallmark of chronic renal failure. TGF-β has been implicated in the chronic progression of IgA glomerulonephritis and diabetes-related nephropathy[21-23]. Furthermore, the expression of the TGF-β/Smad signaling pathway has been reported in children with IgA nephropathy, underscoring its significance in the disease process[20].
TGF-β-induced epithelial-mesenchymal transition, a process in which epithelial cells undergo a transition to a mesenchymal phenotype, plays a vital role in renal fibrosis[24]. Latent transforming growth factor beta binding protein 4 (LTBP4), a regulator of TGF-β, has also been implicated in the pathogenesis of renal fibrosis[25].
Importantly, TGF-β/Smad inhibitors have demonstrated the potential to alleviate the progression of renal fibrosis, highlighting the therapeutic potential of targeting this pathway in the management of uremia[26-28].
Regulatory T Cells and Pulmonary Fibrosis
Pulmonary fibrosis, a progressive and debilitating lung disease, is characterized by the scarring and stiffening of lung tissue, leading to impaired respiratory function. Symptoms such as shortness of breath, dry cough, fatigue, and weight loss can significantly impact the quality of life of affected individuals. Complications like pulmonary hypertension, respiratory failure, and pneumothorax can further exacerbate the condition.
The underlying causes of pulmonary fibrosis are diverse, ranging from environmental pollutants and certain medications to autoimmune disorders, viral infections, and interstitial lung diseases. However, in many cases, the cause remains unknown, a condition termed idiopathic pulmonary fibrosis.
Chronic inflammation is believed to be a common pathway leading to lung fibrosis, and TGF-β has been identified as a key mediator in the pathogenesis of this condition. TGF-β has been detected at sites of extracellular matrix gene expression in human pulmonary fibrosis, suggesting its involvement in the fibrotic process[29].
Acute respiratory distress syndrome (ARDS), a potentially life-threatening condition that can occur after viral or bacterial infections, has been associated with the development of pulmonary fibrosis. Notably, TGF-β has been implicated in the pathophysiology of ARDS, further underscoring its role in lung fibrosis[30,31].
Inhibition of TGF-β receptor signaling has been shown to prevent lung fibrosis in animal models, highlighting the potential therapeutic value of targeting this pathway. Additionally, microRNAs such as miR-133a, which inhibits TGF-β1-induced myofibroblast differentiation, have demonstrated the ability to prevent pulmonary fibrosis in mice models[32].
Regulatory T cells, through their production of TGF-β, play a vital role in the pathogenesis of pulmonary fibrosis. Inhibition of miR-182-5p, which attenuates pulmonary fibrosis via the TGF-β/Smad pathway, further supports the involvement of Tregs in this debilitating condition[33].
The Interplay of End-Organ Damage: Heart, Kidney, Lung, and Liver
The role of regulatory T cells in the pathogenesis of end-organ failure extends beyond individual organ systems, as evidenced by the intricate interplay between the heart, kidneys, lungs, and liver.
Cardiorenal syndrome, a clinical term that describes the co-existence of heart failure and renal failure, highlights the interconnectedness of these organ systems. Similarly, hepatorenal syndrome refers to the concurrent presence of liver failure and renal failure, while the term "hepatocardiorenal syndrome" encompasses the interactions among all three organs[34].
Notably, in cardiorenal syndrome, regulatory T cell populations have been found to be elevated compared to patients with chronic kidney disease alone[35]. This observation suggests that Treg cells may act as key mediators in the interaction between end-organ damage processes.
Furthermore, the concept of cardio-pulmonary-renal interaction underscores the intricate relationships among these vital organs, and the potential for regulatory T cells to contribute to the progression of multi-organ dysfunction[36].
Therapeutic Implications and Conclusion
The enigmatic role of regulatory T cells in organ fibrosis and failure presents both challenges and opportunities for therapeutic intervention. By elucidating the common pathophysiological mechanisms underlying conditions such as systemic sclerosis, heart failure, liver cirrhosis, uremia, and pulmonary fibrosis, we gain invaluable insights into potential therapeutic targets.
Regulatory T cells, through their production of TGF-β, emerge as key mediators of end-stage organ fibrosis. Consequently, strategies aimed at controlling chronic inflammation and the associated tolerable immune reactions may hold the key to preventing and managing these debilitating conditions.
Therapies targeting the TGF-β signaling pathway, such as TGF-β inhibitors or modulators of the associated signaling cascades, have shown promise in preclinical and clinical studies. Furthermore, the regulation of microRNAs and non-coding RNAs that interact with the TGF-β pathway presents a novel therapeutic avenue.
Ultimately, by unraveling the intricate interplay between regulatory T cells, TGF-β, and organ fibrosis, we may unlock the potential to develop effective treatments that can alleviate the burden of these devastating conditions and improve the quality of life for countless individuals worldwide.
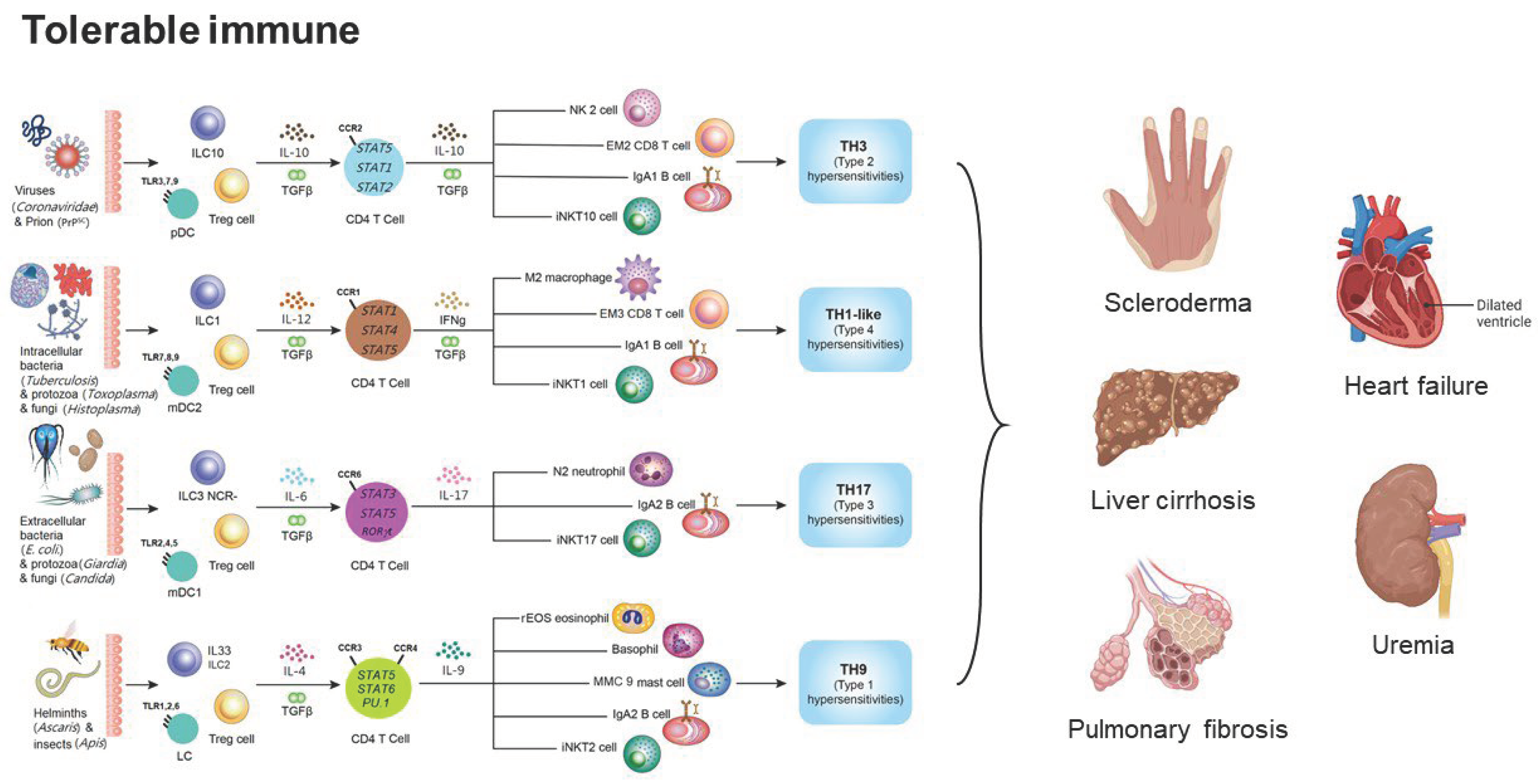
Figure 1.
The framework of tolerable immune responses and their relations to scleroderma, heart failure, liver cirrhosis, uremia, and pulmonary fibrosis. The tolerable immune responses are initiated by Treg cells and can be further divided into TH3, TH1-like, TH17, and TH9 immunological pathways. TH3 tolerable immune reaction is the immune response against viruses and prions. TH1-like tolerable immune reaction is the immune response against intracellular bacteria, protozoa, and fungi. TH17 tolerable immune reaction is the immune response against extracellular bacteria, protozoa, and fungi. TH9 tolerable immune reaction is the immune response against parasites.
Figure 1.
The framework of tolerable immune responses and their relations to scleroderma, heart failure, liver cirrhosis, uremia, and pulmonary fibrosis. The tolerable immune responses are initiated by Treg cells and can be further divided into TH3, TH1-like, TH17, and TH9 immunological pathways. TH3 tolerable immune reaction is the immune response against viruses and prions. TH1-like tolerable immune reaction is the immune response against intracellular bacteria, protozoa, and fungi. TH17 tolerable immune reaction is the immune response against extracellular bacteria, protozoa, and fungi. TH9 tolerable immune reaction is the immune response against parasites.
References
- Chu, Y.T.; Liao, M.T.; Tsai, K.W.; Lu, K.C.; Hu, W.C. Interplay of Chemokines Receptors, Toll-like Receptors, and Host Immunological Pathways. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Hu, W.C. A Framework of All Discovered Immunological Pathways and Their Roles for Four Specific Types of Pathogens and Hypersensitivities. Front Immunol 2020, 11, 1992. [Google Scholar] [CrossRef]
- Wen, T.H.; Tsai, K.W.; Wu, Y.J.; Liao, M.T.; Lu, K.C.; Hu, W.C. The Framework for Human Host Immune Responses to Four Types of Parasitic Infections and Relevant Key JAK/STAT Signaling. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tsai, K.W.; Lu, K.C.; Shih, L.J.; Hu, W.C. Cancer as a Dysfunctional Immune Disorder: Pro-Tumor TH1-like Immune Response and Anti-Tumor THalphabeta Immune Response Based on the Complete Updated Framework of Host Immunological Pathways. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J Exp Med 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Ayers, N.B.; Sun, C.M.; Chen, S.Y. Transforming growth factor-beta signaling in systemic sclerosis. J Biomed Res 2018, 32, 3–12. [Google Scholar] [CrossRef] [PubMed]
- McCormick, L.L.; Zhang, Y.; Tootell, E.; Gilliam, A.C. Anti-TGF-β Treatment Prevents Skin and Lung Fibrosis in Murine Sclerodermatous Graft-Versus-Host Disease: A Model for Human Scleroderma. The Journal of Immunology 1999, 163, 5693–5699. [Google Scholar] [CrossRef] [PubMed]
- D'Amico, F.; Skarmoutsou, E.; Marchini, M.; Malaponte, G.; Caronni, M.; Scorza, R.; Mazzarino, M.C. Genetic polymorphisms of FOXP3 in Italian patients with systemic sclerosis. Immunol Lett 2013, 152, 109–113. [Google Scholar] [CrossRef]
- Tang, J.; Lei, L.; Pan, J.; Zhao, C.; Wen, J. Higher levels of serum interleukin-35 are associated with the severity of pulmonary fibrosis and Th2 responses in patients with systemic sclerosis. Rheumatol Int 2018, 38, 1511–1519. [Google Scholar] [CrossRef]
- Edgley, A.J.; Krum, H.; Kelly, D.J. Targeting fibrosis for the treatment of heart failure: a role for transforming growth factor-beta. Cardiovasc Ther 2012, 30, e30–e40. [Google Scholar] [CrossRef]
- Lim, H.; Zhu, Y.Z. Role of transforming growth factor-beta in the progression of heart failure. Cell Mol Life Sci 2006, 63, 2584–2596. [Google Scholar] [CrossRef]
- Humeres, C.; Shinde, A.V.; Hanna, A.; Alex, L.; Hernandez, S.C.; Li, R.; Chen, B.; Conway, S.J.; Frangogiannis, N.G. Smad7 effects on TGF-beta and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Chang, S.L.; Hsiao, Y.W.; Tsai, Y.N.; Lin, S.F.; Liu, S.H.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chao, T.F.; Hu, Y.F. , et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J Mol Cell Cardiol 2018, 122, 69–79. [Google Scholar] [CrossRef]
- Cappuzzello, C.; Di Vito, L.; Melchionna, R.; Melillo, G.; Silvestri, L.; Cesareo, E.; Crea, F.; Liuzzo, G.; Facchiano, A.; Capogrossi, M.C. , et al. Increase of plasma IL-9 and decrease of plasma IL-5, IL-7, and IFN-gamma in patients with chronic heart failure. J Transl Med 2011, 9, 28. [Google Scholar] [CrossRef]
- Romano, A.; Hou, X.; Sertorio, M.; Dessein, H.; Cabantous, S.; Oliveira, P.; Li, J.; Oyegue, S.; Arnaud, V.; Luo, X. , et al. FOXP3+ Regulatory T Cells in Hepatic Fibrosis and Splenomegaly Caused by Schistosoma japonicum: The Spleen May Be a Major Source of Tregs in Subjects with Splenomegaly. PLoS Negl Trop Dis 2016, 10, e0004306. [Google Scholar] [CrossRef]
- Mou, H.; Wu, S.; Zhao, G.; Wang, J. Changes of Th17/Treg ratio in the transition of chronic hepatitis B to liver cirrhosis and correlations with liver function and inflammation. Exp Ther Med 2019, 17, 2963–2968. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Sawada, S.; Sato, Y.; Nakanuma, Y. Naturally-occurring regulatory T cells are increased in inflamed portal tracts with cholangiopathy in primary biliary cirrhosis. J Clin Pathol 2007, 60, 1102–1107. [Google Scholar] [CrossRef]
- Elhani, I.; Pillebout, E.; Terrier, B.; Hankard, A.; Vrtovsnik, F.; Jourde-Chiche, N.; Greillier, S.; Groh, M.; Belfeki, N.; Bigot, A. , et al. IgA Vasculitis With Underlying Liver Cirrhosis: A French Nationwide Case Series of 20 Patients. J Rheumatol 2021, 48, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ren, P.; Wang, Y.; Feng, S.; Wang, C.; Shen, X.; Weng, C.; Lang, X.; Chen, Z.; Jiang, H. , et al. Serum Matrix Metalloproteinase-7 Level is Associated with Fibrosis and Renal Survival in Patients with IgA Nephropathy. Kidney Blood Press Res 2017, 42, 541–552. [Google Scholar] [CrossRef]
- Wu, W.; Jiang, X.Y.; Zhang, Q.L.; Mo, Y.; Sun, L.Z.; Chen, S.M. Expression and significance of TGF-beta1/Smad signaling pathway in children with IgA nephropathy. World J Pediatr 2009, 5, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Niemir, Z.I.; Stein, H.; Noronha, I.L.; Kruger, C.; Andrassy, K.; Ritz, E.; Waldherr, R. PDGF and TGF-beta contribute to the natural course of human IgA glomerulonephritis. Kidney Int 1995, 48, 1530–1541. [Google Scholar] [CrossRef]
- Border, W.A.; Noble, N.A. Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int 1998, 54, 1390–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zou, Y.; Liu, F. Transforming Growth Factor-Beta1 in Diabetic Kidney Disease. Front Cell Dev Biol 2020, 8, 187. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Duan, J.; Li, H.; Wang, Y.; Wang, F.; Chu, J.; Sun, J.; Liu, M.; Wang, C.; Lu, C. , et al. Hydroxysafflor Yellow A Ameliorates Renal Fibrosis by Suppressing TGF-beta1-Induced Epithelial-to-Mesenchymal Transition. PLoS One 2016, 11, e0153409. [Google Scholar] [CrossRef]
- Su, C.T.; See, D.H.W.; Huang, Y.J.; Jao, T.M.; Liu, S.Y.; Chou, C.Y.; Lai, C.F.; Lin, W.C.; Wang, C.Y.; Huang, J.W. , et al. LTBP4 Protects Against Renal Fibrosis via Mitochondrial and Vascular Impacts. Circ Res 2023, 133, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yue, S.; Fang, J.; Zeng, J.; Chen, S.; Tian, J.; Nie, S.; Liu, X.; Ding, H. MicroRNA-10a/b inhibit TGF-beta/Smad-induced renal fibrosis by targeting TGF-beta receptor 1 in diabetic kidney disease. Mol Ther Nucleic Acids 2022, 28, 488–499. [Google Scholar] [CrossRef]
- Feng, J.; Xie, L.; Kong, R.; Zhang, Y.; Shi, K.; Lu, W.; Jiang, H. RACK1 silencing attenuates renal fibrosis by inhibiting TGF-beta signaling. Int J Mol Med 2017, 40, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhou, W.; Yao, F.; Song, J.; Xu, Y.; Deng, Z.; Diao, H.; Li, S. MicroRNA-302b mitigates renal fibrosis via inhibiting TGF-beta/Smad pathway activation. Braz J Med Biol Res 2021, 54, e9206. [Google Scholar] [CrossRef]
- Hou, Z.; Ye, Q.; Qiu, M.; Hao, Y.; Han, J.; Zeng, H. Increased activated regulatory T cells proportion correlate with the severity of idiopathic pulmonary fibrosis. Respir Res 2017, 18, 170. [Google Scholar] [CrossRef]
- Hu, W.; Yen, Y.T.; Singh, S.; Kao, C.L.; Wu-Hsieh, B.A. SARS-CoV regulates immune function-related gene expression in human monocytic cells. Viral Immunol 2012, 25, 277–288. [Google Scholar] [CrossRef]
- Shih, L.J.; Yang, C.C.; Liao, M.T.; Lu, K.C.; Hu, W.C.; Lin, C.P. An important call: Suggestion of using IL-10 as therapeutic agent for COVID-19 with ARDS and other complications. Virulence 2023, 14, 2190650. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-beta1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Q.; Zhou, Y.; Yang, Z.; Tan, M. Inhibition of miR-182-5p attenuates pulmonary fibrosis via TGF-beta/Smad pathway. Hum Exp Toxicol 2020, 39, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Kazory, A.; Ronco, C. Hepatorenal Syndrome or Hepatocardiorenal Syndrome: Revisiting Basic Concepts in View of Emerging Data. Cardiorenal Med 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Duni, A.; Kitsos, A.; Bechlioulis, A.; Markopoulos, G.S.; Lakkas, L.; Baxevanos, G.; Mitsis, M.; Vartholomatos, G.; Naka, K.K.; Dounousi, E. Differences in the Profile of Circulating Immune Cell Subsets in Males with Type 2 Cardiorenal Syndrome versus CKD Patients without Established Cardiovascular Disease. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Husain-Syed, F.; McCullough, P.A.; Birk, H.W.; Renker, M.; Brocca, A.; Seeger, W.; Ronco, C. Cardio-Pulmonary-Renal Interactions: A Multidisciplinary Approach. J Am Coll Cardiol 2015, 65, 2433–2448. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



