
Submitted:
10 April 2025
Posted:
11 April 2025
You are already at the latest version
Abstract
The growing human population increases the need for food, beyond what terrestrial sources can provide. This boosts aquaculture demand for molluscs, fish, and crustaceans. Molluscs are popular for their nutritional benefits, making them a profitable industry. Despite a 3% annual growth in mollusc populations, recent high mortality rates and population losses due to poor feeding practices and water pollution have made them more disease prone. Limited treatment options exist for mollusc diseases in aquaculture systems. Hence, developing rapid, sensitive, and cost-effective diagnostic tools for field use is essential to identify and prevent infections promptly. Recently developed isothermal nucleic acid amplification technologies, like loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA), offer rapid results within an hour. This review examines these isothermal diagnostic techniques for mollusc pathogens and their potential for field application.
Keywords:
molluscs
; pathogens
; isothermal amplification
; point-of-care testing
1. Introduction
The human population is steadily increasing, with the global population recently surpassing eight billion people [1]. This growth escalates the demand for food production, which terrestrial-based meats cannot fulfil alone. Aquacultural products provide consumers with various protein sources, such as finfish, crustaceans, and molluscs. Commercial molluscs can include bivalve molluscs (mussels, clams, scallops, cockles, and oysters), cephalopods (octopus, squid and cuttlefish), and gastropods (abalone and conch) [2,3]. Molluscs represent 11% of the worldwide seafood trade, with over half of this comprising squids, cuttlefish and octopus [3,4]. Molluscs are a good source of omega-3 fatty acids, iron, selenium, and zinc, while having lower levels of carbohydrates and fats compared to land-based proteins, making them a healthier dietary option than terrestrial meat [5,6]. Molluscs’ importance is not only limited to their nutritional and commercial value but they also play a critical role in maintaining the stability of the ocean's ecosystem (Eutrophication) as they filter phytoplankton (completed mainly by bivalves) and help in carbonate buffering mechanisms [2,5]. Furthermore, the inedible shells of molluscs are used in medicine, cosmetics, jewelry, and chemicals industries such as dyes and catalysts [2,5,6,7].
Despite the global COVID-19 pandemic's impact on international trade, the production of molluscs has continued to grow (Figure 1A). Since 1990, mollusc production (excluding cephalopods) has seen an annual growth rate of 2.7%, with a million ton increase in farmed molluscs from 2020 to 2022 (Figure 1A) [3,4]. In 2018, mollusc farming generated USD 34.6 billion in revenue, second only to finfish production, which brought in USD 139.7 billion [3]. Mollusc consumption reached an average of 3.1 kg per person in 2017 and still accounted for half of all shellfish consumption in 2022 [3,4]. In 2018, the production of shelled molluscs from marine and coastal aquaculture was 17.3 million tons (56.2%), which exceeded that of finfish and crustaceans, with production volumes of 7.3 million tons, and 5.7 million tons, respectively [3].
Asia is the world's largest producer of molluscs, with an annual production of 17,449,826 tons in 2022, far surpassing Europe (598,680 tons), followed by America, Oceania, and Africa [4]. Within Asia, China is the leading producer, generating approximately 16 million tons of highly commercial molluscs such as mussels, squid, cuttlefish, and abalone, with 75% of the world's molluscs being farmed in 2022 [4] (Figure 1B).
The intensive expansion of aquatic farming, combined with environmental pollution and the natural absence of adaptive immunity in molluscs, makes cultivated mollusc species highly susceptible to disease. This vulnerability can lead to significant production losses when infectious disease outbreaks occur [8]. In a recent 2018 census study of aquaculture, performed by the Department of Agriculture in the USA, diseases were listed as the main cause of aquaculture production losses [3]. Molluscs are susceptible to many viral [9,10], bacterial [11], parasitic [12,13], and fungal diseases [14], causing mortality or slow growth rates that have detrimental impacts on the domestic and international markets. Identifying infectious pathogens quickly is crucial for sustaining the mollusc production industry.
2. Pathogens of High Biosecurity Concern in Global Mollusc Production and Their Current Molecular Diagnostic Methods
There have been significant outbreaks of various diseases on mollusc farms worldwide, some of which have resulted in 100% mortality rates. Most mollusc diseases are transmitted through direct contact between infected animals or by exposure to contaminated water [15]. Despite strict regulations on the trade of molluscs, infectious diseases continue to spread both among different species and within the same species globally [16,17,18,19]. The 2024 World Organization for Animal Health (WOAH) list of notifiable diseases includes five parasitic pathogens that have severe effects on molluscs globally: Bonamia ostreae, Bonamia exitiosa, Marteilia refringens, Perkinsus marinus, and Perkinsus olseni, along with abalone herpesvirus. Since clinical signs are not always present to confirm infection, diagnostic methods are essential to inform practices on controlling the spread of pathogens on farms and during the transportation of molluscs between countries. This summary outlines the current molecular detection methods used to identify mollusc diseases (Table1).
2.1. Viral Diseases Affecting Molluscs
2.1.1. Abalone Viral Ganglioneuritis (AVG)
The abalone production industry is under significant threat from the abalone herpesvirus (AbHV), also known as Haliotid herpesvirus-1 (HaHV-1) in Australia [20]. This neurotropic virus causes ganglioneuritis, leading to the condition termed abalone viral ganglioneuritis (AVG) [21]. The virus is believed to have first emerged in China in the late 1990s and later spread to Taiwan in 2003 [21]. The Taiwan outbreak mainly affected cultured (Haliotis diversicolor supertexta) or small abalone and resulted in losses exceeding USD 11.5 million for the industry [22,23,24]. In 2005, subsequent outbreaks in Victoria, Australia led to 100% mortality in blacklip (Haliotis rubra), greenlip (Haliotis laevigata), and their hybrid due to AVG [25]. Abalone are generally vulnerable to AbHV at all life stages; however, complete resistance has been observed in the New Zealand paua (Haliotis iris) after artificial infection [26]. Additionally, disc abalone (Haliotis discus hannai) has been identified as an asymptomatic carrier of the virus, harboring it without displaying disease symptoms under experimental conditions [20]. Further research is necessary to determine whether H. discus hannai has natural immunity or if environmental factors contribute to the disease manifestation [20]. The AbHV virus is enveloped and icosahedral, measuring 100 nm, and is classified under the genus Aurivirus within the family Malacoherpesviridae and order Herpesvirales [27]. AVG is cytocidal, impacting the nervous system and causing tissue necrosis associated with the nerves [28]. Infected abalone during the Taiwan outbreak exhibited symptoms such as mantle recession and muscle atrophy, while Victorian abalone displayed symptoms like swollen mouths and prolapsed odontophores [24,28]. The variation in disease presentation among different abalone species complicates the diagnosis of pathogen infections based solely on clinical signs [21].
Due to the virus's severity and rapid spread, prompt and accurate on-farm detection is crucial. However, the absence of a gold standard detection recommendation from the WOAH complicates matters. Corbeil et al. (2010) developed a TaqMan quantitative PCR (qPCR) assay capable of detecting fewer than 300 copies of a recombinant plasmid containing the target open reading frame (ORF) 38 (also known as 49) of the AbHV gene. The emergence of new AbHV-1 strains in Tasmania necessitated the development of new qPCR assays targeting ORF 66 and 77 [15,29,30]. These new assays can detect all known strains of HaHV-1 in Australia, even during sub-clinical stages, but require extraction of the nerves making them costly and time consuming assays [29]. With the emergence of new variants and the high susceptibility of most abalone species to this virus, establishing a robust surveillance system, particularly in low-resourced laboratories, is essential. As there is currently no vaccine available against AbHV, quick and accurate diagnosis is vital for containing the virus before outbreaks occur.
2.1.2. Abalone Shrivelling Syndrome (AbSS)
The inaugural documentation of Abalone Shrivelling Syndrome (AbSS) traces back to 1999 in China, predominantly impacting small abalone [31,32]. Researchers identified a chimeric, double-stranded DNA virus in afflicted specimens, subsequently christened the abalone shrivelling syndrome-associated virus (AbSV). The genome of this viral pathogen shares extensive homology with both bacteriophages and bacteria, suggesting a complex ancestry [33]. Post its initial emergence, AbSS has proliferated beyond China, reaching Taiwan and other nations, thereby affecting abalones across various developmental stages. The syndrome manifests through several clinical signs, including diminished appetite, pedal disc muscle wastage, mantle darkening, and a propensity for the abalones to dislodge from their reef habitats, often resulting in mortality post-detachment [34,35,36]. The most recent significant outbreak of AbSS was recorded in China in 2005. Despite the continued presence of the pathogen, there has been a noticeable decrease in its pathogenicity. This decline is potentially attributable to advancements in aquaculture practices, thereby enhancing survival rates. Additionally, observed mutations within the AbSV genome may have reduced the virus's lethality [22,37].
To detect AbSS, nested PCR (nPCR) and qPCR assays have been developed, offering high levels of sensitivity and specificity. However, these methods require the dissection of animal mantles and feet for sampling, necessitating specialist expertise. Notably, the qPCR assay, which targets ORF2 of AbSS, can identify as few as 10 copies of the recombinant plasmid that harbors the target gene, highlighting the assay's precision [34,35]. Despite this, molecular analyses of moribund abalones from Taiwan have shown a high genetic congruence with AbSV, yet in situ hybridization (ISH) has not successfully linked these cases to AbSS, indicating a discrepancy in detection methodologies [31]. This disparity accentuates the imperative for developing accurate, swift, and easily deployable diagnostic tools capable of identifying the virus and its mutant strains.
2.1.3. Acute Viral Necrobiotic Disease
The acute viral necrobiotic disease is caused by the acute viral necrobiotic virus (AVNV), which first appeared in China in the 1980s, leading to significant deaths among the zhikong scallop, Chlamys farreri [38]. AVNV is characterized by its spiked spherical envelope morphology containing DNA genomic materials, measuring approximately 170 nm in diameter [39]. The virus primarily targets the host's epithelial cells and connective tissues, resulting in necrosis of essential organs such as the gills, mantles, intestine, and digestive gland [39,40]. The virus predominantly affects immature scallops around two years old, with infection rates peaking during the summer months when temperatures are between 25-27°C. The mortality rate associated with AVNV infections is alarmingly high, estimated between 60-90%, and fatalities typically occur within 2-9 days post-infection [41,42].
Various techniques, including serological, microscopic, and molecular detection assays, have been developed to identify AVNV. Fluorescence quantitative PCR has demonstrated the ability to detect low numbers of the virus with high specificity [42,43,44,45]. However, despite advancements in rapid detection methods, none have been optimized for field application, as they primarily rely on aseptic isolation of gills and mantle tissue, which is challenging to perform on-site [44]. This highlights a critical gap in current diagnostic capabilities and emphasizes the need for further research in this area.
2.1.4. Infection with Ostreid Herpesvirus -1
The first documented case of ostreid herpesvirus-1 (OsHV-1) infection was identified in 1972 in the United States [16]. Despite extensive research, the definitive origin species responsible for transmitting OsHV-1 remains unknown. The virus has been detected in a variety of bivalve species, including oysters, mussels, clams, and scallops [16,21]. The virus's ability to infect such a broad range of molluscs may be due to the co-cultivation of different aquatic organisms, which typically do not coexist, facilitating viral transmission. Another possibility is that OsHV-1 could be a mutation of another virus that originally infected a specific bivalve species before evolving to infect more broadly [17]. New variants of OsHV-1 continue to emerge, with the most recent being OsHV-1 µvar, which is characterized by a deletion of approximately 2.8 kbp from the original 167.8 kbp reference strain [46]. The µvar variant first appeared in France and is recognized as the most virulent form of the virus, devastating all life stages of the Pacific oyster in 2008, and subsequently spreading to Europe, Asia, New Zealand, and Australia, eventually being included on the WOAH list in 2013 [22,47,48]. Infected larvae often show reduced food intake and general activity, leading to increased mortality, while adult oysters display nonspecific symptoms like sluggish behavior and gapping of the shell [33,49].
Efforts to isolate the virus through cell culture techniques have been unsuccessful, though infectivity has been established using filtrates from homogenized infected tissues [50]. Detection methods for OsHV-1 include qualitative and quantitative PCR, ISH, and immunochemistry techniques [49,51,52,53,54]. However, these methodologies are impractical for routine surveillance in-field due to their high cost, labor-intensive procedures, and reliance on specialized laboratory equipment and trained personnel for gonad and mantle dissection [51]. The WOAH recommends qualitative and quantitative PCR followed by sequencing for the confirmation of infections [33]. Given the lack of specific symptoms and the aggressive nature of OsHV-1, there is a pressing need for reliable, rapid, and cost-effective, point-of-care detection methods.
2.2. Parasitic Diseases Affecting Molluscs
2.2.1. Haplosporidiosis
The phylum Haplosporidia encompasses around 36 species of endoparasites that impact marine and freshwater invertebrates. This phylum includes Urosporidium, Haplosporidium, Minchinia taxa, and recently added Bonamia [55]. A significant pathogen within this group is Haplosporidium spp., which leads to haplosporidiosis in Pacific oysters, Crassostrea gigas, across the USA, Europe, and Asia [56]. Haplosporidium nelsoni, commonly referred to as MSX, is the most extensively studied member of this genus. It was first identified in Delaware Bay in the 1950s, originally named Minchinia nelson, and is known for causing severe mortalities in eastern oysters, C. virginica [57]. H. nelsoni triggered a significant outbreak in oyster farms in the USA, culminating in 90% mortalities [58]. The impact of this outbreak led to the establishment of annual Oyster Mortality Conferences to investigate the issue throughout the following decade [59]. This disease is most prevalent in high salinity waters, where the parasite damages the epithelial cells of the oysters' digestive tubules, ultimately leading to their death. Notably, higher mortality rates are observed in the summer months, particularly when the water is at around 20 ppt salinity [60]. Another species that causes haplosporidiosis is Haplosporidium costale (SSO), which also targets eastern oysters and is morphologically similar to H. nelsoni.
Traditional morphological detection methods do not allow for differentiation between these two species, but certain molecular techniques like conventional PCR (cPCR) and ISH can identify Haplosporidia spp. individually [61]. Penna et al. (2001) developed a multiplex PCR (mPCR) to detect three pathogens, H. nelsoni, H. costale, and Perkinsus marinus, for effective and rapid pathogen detection. However, despite the availability of numerous successful detection techniques, a quick and economical method that does not require trained specialists for tissue extraction and test execution remains is lacking [62,63].
2.2.2. Bonamiosis
Bonamiosis, also known as Microcell disease, is caused by several members of the genus Bonamia. These small (2-3 µm) intracellular protozoa mainly infect hemocytes, but they can also be found in some extracellular tissues and may become systemic in severe cases [15,64,65]. Oysters are particularly vulnerable to this disease in waters that are cold and have high salinity. As a result, diseased oysters can experience mass mortality, with levels reaching 90%, which significantly impacts oyster populations and raises concerns about maintaining a balanced ecosystem, especially in Europe [66,67,68]. Currently, four members of Bonamia are known: B. exitiosa, B. ostreae, B. roughley, and B. perspora. B. roughley causes Australian winter disease, affecting Sydney rock oysters, Saccostrea glomerata, in southeast Australia [67]. The newly described B. perspora has been found in Ostrea equestris and O. lurida [67]. B. ostreae was first detected in the flat oyster species O. edulis in the 1970s in the USA, with serious consequences for the oyster industry in the northern hemisphere [66,69,70,71,72]. B. exitiosa was initially reported in New Zealand in 1985, causing severe damage to O. chilensis production, with a significant decrease in populations by 91% in 1990 [70,73,74,75]. Although oysters from the genus Ostrea were thought to be the only hosts for Bonamia spp., recent studies have shown that oysters from the genera Crassostrea, Saccostrea, and Dendostrea are also susceptible [66,68]. Infected hosts may show no symptoms or exhibit common sickness signs, such as ulcers, eroded or discolored gills, and poor condition, which may lead to mortality [15,68]. Interestingly, some populations of O. edulis show better tolerance to the parasite, possibly due to low parasite levels or partial host resistance from long-term exposure [76]. Despite various attempts to clean and reuse previously infested waters to cultivate the oysters, the pathogen could not be eradicated [72,77]. Mortality peaks when infections coincide with Marteilia refringens or other Bonamia species. For example, dual infection with B. ostreae and M. refringens reduced flat oyster annual production in Europe from 29,595 tons to 5,921 tons between 1961 and 2000, highlighting the need for specific detection methods to distinguish infection causes [66,73].
Various molecular biology techniques have been developed to detect Bonamia spp., including ISH, cPCR, qPCR, and mPCR; however, none of these methods are field-deployable, as they require gill tissue to be excised. This limitation poses significant challenges in real-time detection, especially in remote areas where immediate access to laboratory facilities is often not feasible [18,78,79,80]. Histology remains the gold standard for detection, but it is challenging to differentiate between Bonamia species using this method, especially in early infection stages [3]. Specific ISH assays can detect closely related B. exitiosa–B. roughley clade successfully [81]. The PCR- restriction fragment length polymorphism (RFLP) technique can differentiate polymorphisms among Bonamia species, yet it remains labor-intensive for field use [73,82]. A TaqMan qPCR assay designed to detect the internal transcribed spacer (ITS) sequence of Bonamia spp. has demonstrated faster and more sensitive results than histology and, it offers similar sensitivity to previously developed cPCR methods targeting the small sub-unit ribosomal DNA (SSU rDNA) [79,80]. Currently, the primary strategy for reducing infections involves cultivating lighter-weight oysters and minimizing environmental stresses that could increase their susceptibility to disease, underscoring the urgent need for developing point-of-care tests for early detection [83].
2.2.3. Marteiliosis
Marteiliosis poses a significant threat to molluscan populations, caused by protozoan parasites from the phylum Cercozoa and order Paramyxida [84]. This disease is linked to several species within the genus Marteilia, such as M. sydneyi, M. granula, M. lenghei, and others [85,86]. These parasites have led to considerable production declines in mussel, oyster, clam, and cockle farms globally [12,18,87,88,89]. Notably, marteiliosis can result in mass mortalities (50-90%) among adult oysters in their second year of infection [12,18,87,88,89]. M. sydneyi is particularly significant, primarily affecting the Sydney rock oyster [18,90,91,92]. The WOAH recognized this pathogen's significance in the 1990s due to its effects on bivalve mollusc populations [90]. M. refringens was first discovered in the 1960s in flat oysters, and since then, infections have been reported in various life stages of other aquatic animals across Europe [18,91,93]. Recently, M. refringens has been classified into M and O types, specifically affecting mussels and oysters [18,91,94]. While the lifecycle of Marteilia spp. remains partly unclear, it has been established that the parasite follows an indirect life cycle as other marine organisms are involved. However, the parasite can survive up to three weeks outside an animal body in suitable conditions [93,95]. The pathology of marteiliosis differs with the specific pathogen and host, usually leading to reduced growth rates, poor health, tissue necrosis, and body shrinkage in infected molluscs [95,96,97,98,99]. In advanced stages, the accumulation of parasites in the digestive gland can lead to starvation and death [95,96,97,98,99]. Efforts to breed more resistant oyster species have faced challenges due to the wide susceptibility among various species; however, the Pacific oyster shows some resistance [12].
Detection and identification of Marteilia species have traditionally relied on histological methods, but molecular techniques are now more effective for species-level identification. The gold standard for detecting marteiliosis remains histology, often confirmed with ISH assays to validate findings [100]. PCR techniques, including generic PCR and nPCR assays, have been developed to detect various Marteilia species and differentiate between types, providing quicker identification methods [91,94]. Recently, mPCR assays have been introduced to detect pathogens from both Marteilia and Bonamia genera; however, these methods still require invasive tissue extraction, which is not ideal for field detection [18].
2.2.4. Marteilioides
Marteilioides, a disease first identified in the 1970s within a Korean farming operation of Pacific oysters, poses a significant threat to aquaculture due to its pathogenic effects on oyster populations [68,101]. Infected specimens initially displayed yellowish, spherical nodules on the mantle, which were mistakenly thought to be amoebic infections [101]. However, further investigations using transmission electron microscopy (TEM) clarified this misidentification, leading to the classification of the new genus Marteilioides [101,102]. Among the species in this genus, Marteilioides chungmuensis has particularly harmful effects on the oysters' reproductive systems, targeting the oocyte's cytoplasm, obstructing egg release from the ovarian follicle, and causing infertility. This then led to a significant decline in seed oyster production, adversely impacting the aquaculture industries in Korea and Japan [68,103,104]. Additionally, M. branchialis has been linked to infections in the gills of Sydney rock oysters, with pathogenic severity increasing when co-infected with M. sydneyi, resulting in higher mortality rates [105].
A study assessing the sensitivity and specificity of current detection methods for Marteilioides spp. found that ISH showed greater sensitivity compared to histology alone [106]. The ISH assay identified several immature parasites in the early development stages. Although PCR demonstrated high sensitivity, inconsistencies with histology and ISH led to many samples being incorrectly classified as false positives or negatives [106]. Despite ISH's superior performance over histology and PCR, its labor-intensive and time-consuming nature, along with the requirement for specialists to aseptically dissect the animal for sampling, remains a significant disadvantage. These factors limit its practicality as a point-of-care detection method, particularly in urgent situations where rapid diagnosis is essential.
2.2.5. Denman Island Disease
Denman Island recorded the first observed case of the disease in 1960, which primarily occurs when temperatures fall below 10°C [68]. The causative agent is the intracellular protistan parasite, Mikrocytos mackini. There is limited information regarding its morphology, genomic DNA, and host interactions [107]. Denman Island disease affects various oyster species across multiple countries [67,75]. Older oysters are generally more severely affected; symptoms include hemocyte infiltration and necrosis, leading to mortality rates of approximately 30% [68]. The disease was previously on the WOAH notifiable diseases list and is currently included as an exotic disease in European (EU) legislation updated in 2018 [67]. A particularly intriguing characteristic of M. mackini is its lack of mitochondria, complicating efforts to determine its evolutionary position [107]. Further genetic studies have indicated that M. mackini has evolutionarily reduced mitochondrial-related organelles (MROs) known as mitosomes, which replace traditional mitochondria [107].
Significant advancements in detecting this parasite have been made with developing sensitive and specific assays, including cPCR and fluorescent situ hybridization (FISH), targeting the SSU rDNA of M. mackini [108]. Additionally, an undefined qPCR assay was created to target the ITS-2 region in the rDNA of M. mackini. This assay can detect as few as 2-5 copies of genomic DNA from samples collected from the mid-body cross-section of oysters, which is notably more effective than the poorer DNA extraction results obtained from the mantle or adductor muscle [109]. However, the variability in results from samples taken from different parts of the oyster highlights the challenges involved in this process. This inconsistency underscores the need for expertise in sampling techniques to ensure accurate and reliable detection, as different anatomical regions may yield different quality and quantity of DNA.
2.2.6. Perkinsosis
The genus Perkinsus comprises various species that infect hosts through direct contact, causing a condition known as perkinsosis. This disease can lead to numerous detrimental effects on host organisms, such as severely retarded growth, behavioral changes, inflammation, necrosis, and a significant decline in physiological functions like growth, gonadal maturation, reproduction, and immunocompetence. These adverse effects can lead to mortality rates as high as 95% after a year of infection, particularly when water temperatures rise above 20°C [68,110,111,112,113]. Several Perkinsus species have been documented, including P. atlanticus, P. qugwadi, P. andrewsi, P. chesapeaki, P. mediterraneus, P. honshuensis, P. beihaiensis, P. marinus, and P. olseni [114,115,116]. However, only a subset of these species meets the recognized criteria for classification within the genus. For example, P. andrewsi and P. atlanticus have been identified as synonymous with P. chesapeaki and P. olseni, respectively. Conversely, P. qugwadi exhibits distinct phenotypic traits that do not align with established diagnostic markers of the genus, such as its reaction to Ray’s fluid thioglycolate medium (RFTM) [117]. P. marinus was first observed in Mexico during the 1940s in eastern oysters causing perkinsosis disease in some oysters and clams in Europe and North America [118,119]. P. olseni was first reported in Australia in 1972, affecting the blacklip abalone. A recent outbreak of P. olseni in Australia led to a significant drop in abalone production from 300 tons to 94 tons in 2011, causing an annual loss of 500,000 AUD for abalone producers in South Australia since its emergence [115]. The expansive diversity of the Perkinsus genus increases the range of molluscs susceptible to perkinsosis, including oysters, mussels, cockles, clams, and abalone. This broad host range has serious ecological and economic implications, resulting in increased mortality rates in mollusc aquaculture and a decline in the market value of affected species [116,120]. Continued research and documentation of Perkinsus species emphasize the need for ongoing surveillance and mitigation strategies to protect mollusc populations and the aquaculture industry.
The WOAH recommends starting with genus-specific PCR before employing species-specific detection assays for positive samples [15]. Recent advancements in molecular diagnostics have led to the development of genus-specific qualitative and quantitative PCR assays targeting various Perkinsus genes, including ITS, actin, and the ribosomal RNA large subunit (rRNA LSU) gene [121,122,123,124]. A notable innovation is a universal PCR assay that detects a 703 bp sequence unique to the genus Perkinsus, with the exception of P. qugwadi [125]. Given the small size of these parasites, standard histological methods are insufficient for accurate diagnosis, prompting the development of a universal ISH assay targeting the rRNA SSU domain specific to the genus Perkinsus [126]. Species-specific ISH assays have also been created for P. beihaiensis, P. chesapeaki, P. honshuensis, and P. mediterraneus [124,127,128]. In addition, species-specific PCR assays have been designed to accurately identify P. honshuensis, P. chesapeaki, and P. beihaiensis, reflecting ongoing efforts to improve detection methods [124,129,130]. A multiplex PCR- enzyme-linked immunosorbent assay (ELISA) has been developed to identify the intergenic spacer (IGS) sequence of P. marinus, P. atlanticus, and Perkinsus spp., demonstrating sensitivity to as low as 1 pg of DNA, which is 100 times more sensitive than cPCR [131]. Furthermore, a highly sensitive and species-specific qPCR assay for detecting the ITS sequence of P. marinus has been developed, able to detect the pathogen in environmental water samples [125]. However, these remain laboratory-based techniques, with no in-field options available. Species-specific PCR primers can amplify a 455 bp region of the ITS specific to P. olseni’s rRNA gene complex, offering exceptional sensitivity to detect as few as one pathogen cell in 30 mg of tissue however, genomic DNA extraction from excised mantle and gill tissues is a labor-intensive process that requires specialized techniques and expertise, which may not be readily available in farm settings for point-of-care detection [132]. Through these advancements, the landscape of Perkinsus spp. diagnostics continues to evolve, providing refined tools for rapidly detecting this parasite, which is crucial for effective management and control strategies in affected marine environments.

Table 1.
Pathogens affecting commercial molluscs and their susceptible species and current molecular detection methods.
Table 1.
Pathogens affecting commercial molluscs and their susceptible species and current molecular detection methods.
| Pathogen | Susceptible mollusc (s)# | Detection method |
|---|---|---|
| Virus | ||
| Abalone herpesvirus (AbHV) | Blacklip abalone1 Brown abalone2 Disc abalone2 Greenlip abalone1 Pink abalone2 Small abalone1 Tiger abalone1 |
cPCR [133] Sequencing [133] qPCR [29,30] ISH [25,134] |
| Abalone shrivelling syndrome (ASSV) | Disc abalone Small abalone |
qPCR [35] nPCR [34] |
| Acute Viral Necrobiotic Virus (AVNV | Scallops | cPCR [42] qPCR [42,135,136] |
| Ostreid herpesvirus-1 (OsHV-1) | Ark clams Australian flat oyster Bay scallops Blood clam Blue mussels Chilean oyster European clam Flat oyster Great scallop Hairy mussels Manila clam Pacific oyster Portuguese oyster Sydney cockle Sydney rock oysters Telline Virescent Oyster Whelks |
PCR [49] ISH [52,53] qPCR [54] |
| Parasite | ||
| Bonamiaspp. | Australian flat oyster3 Chilean oyster1 Crested oyster1 Dwarf oyster1 European flat oyster1 Hawaiian oyster1 Jinjiang oysters2 Olympia oyster Pacific oyster1 Portuguese oyster1 Suminoe oyster1 Sydney rock oysters |
cPCR [78,79] qPCR [80] mPCR [18] ISH [79,81] |
| Bonamia exitiosa | Argentinian flat oyster Australian flat oyster1 Chilean oyster1 Dwarf oyster1 Eastern oyster European flat oyster1 Olympia oyster Pacific oyster Sydney rock oyster |
qPCR [70] cPCR and sequencing [79,137,138] PCR-RFLP [73] mPCR [70] ISH [79,81,139] |
| Bonamia ostreae | Argentinian flat oyster3 Asiatic oyster Australian flat oyster Chilean oyster European flat oyster1 Pacific oyster3 Portuguese oyster Suminoe oyster1 |
ISH [79,108] qPCR [80,140,141] cPCR [78,79,142] mPCR [70] PCR-RFLP [73] |
| Haplosporidiumspp. | Australian flat oyster Blue mussel California mussel Cockles Eastern oyster European flat oyster Fresh water snails |
qPCR [143,144] cPCR [61,145,146] mPCR [63] ISH [61] |
| Haplosporidium nelson | Eastern oyster Pacific oyster |
ISH [147] cPCR [148] qPCR [149] mPCR [63] |
| Marteiliaspp. | Argentinian flat oyster3 Australian flat oyster Banded Carpet Shell Blacklip oyster Blacklip pearl oyster Blue mussel Calico scallop Chilean oyster Common cockle Dwarf oyster Eastern oyster European flat oyster Grooved razor clam Hooded oyster Iwagaki oyster Jackknife clam Manila clam Maxima clam Mediterranean mussel Northern horse mussel Pacific oyster Palourde clam Peppery furrow shell Pod razor Puelchean Oyster Pullet carpet shell Rock oyster Striped venus clam Suminoe oyster Venerid clam |
cPCR [90,91,94] ISH [94] RFLP-PCR [150] |
|
Marteilia refringens |
Argentinian flat oyster2 Asiatic oyster1 Australian flat oyster2 Banded Carpet Shell Blue mussela1 Calico scallop2 Chilean oyster oyster1 Common cockle1 Dwarf oyster2 Eastern oyster1 European flat oyster1 Grooved razor clam1 Hooded oyster1 Jackknife clam Mediterranean mussel1 Olympia oyster1 Pacific oyster2 Palourde clam Planktonic copepods2 Pod razor Pullet carpet shell Small brown mussel2 Striped venus clam1 |
nPCR [91,151] cPCR and sequencing [90,94,98] mPCR [18] qPCR [152] ISH [90,91,100,153] |
| Marteilia sydneyi | Flat oyster Sydney rock oyster |
cPCR [154] mPCR [18] ISH [155] |
| Marteilioidesspp. | Manila Clam Northern blacklip oyster Pacific oyster Suminoe Oyster |
nPCR [156] |
|
Marteilioides chungmuensis |
Iwagaki oyster Manila clam Pacific Oyster Pacific oyster Suminoe Oyster |
cPCR [103,157] ISH [103] |
| Mikrocytos mackini | Eastern oyster European flat oyster Olympia flat oyster Pacific oyster |
cPCR [158] qPCR [109] ISH [159] FISH [158] |
| Perkinsusspp. | Asian littleneck clam Baltic clam Eastern oyster European flat oyster Hong Kong oyster Mangrove oyster Manila Clam Palourde clam Soft shell clam Stout tagelus Suminoe Oyster Sydney cockle1 Yesso scallop |
cPCR [124,160] ISH [126] PCR—DGGE1 [161] mPCR-ELISA [131] |
| Perkinsusandrewsi | Baltic Clam | cPCR [129] |
| Perkinsusatlanticus | Palourde clam | cPCR [162] mPCR-ELISA [131] |
| Perkinsosis marinus | Baltic macoma Blue mussel Cortez oyster1 Eastern oyster1 Mangrove oyster1 Pacific oyster1 Soft shelled clam Suminoe oyster1 |
cPCR [125,160] ISH# [126,128,163] qPCR [122,125] mPCR-ELISA [131] RFLP-PCR [164] |
| Perkinsosis olseni | Akoya pearl oyster1 Asian littleneck clam1 Australian flat oyster1 Blacklip abalone1 Blacklip pearl oyster1 Crocus clam1 European aurora venus clam1 Giant clam1 Greenlip abalone1 Green-lipped mussel1 Japanese pearl oyster1 Kumamoto oyster Manila clam1 Maxima clam1 New Zealand ark shell1 New Zealand cockle1 New Zealand pauaa1 New Zealand pipia1 New Zealand scallop1 Pacific oyster1 Pearl oyster1 Pullet carpet shell1 Sand cockle Silverlip pearl oyster1 Staircase abalone1 Suminoe oyster1 Sydney cockle1 Venerid clam1 Venerid commercial clam Venus clam Wedge shell Whirling abalone1 |
ISH [126,163,165] cPCR [125,163] qPCR [125] |
1Naturally susceptible, 2Experimentally susceptible, 3 No complete evidence of susceptibility, in situ hybridization (ISH), fluorescent ISH (FISH), polymerase chain reaction (PCR), conventional PCR (cPCR), nested PCR (nPCR), quantitative PCR (qPCR), multiplex PCR (mPCR), enzyme-linked immunosorbent assay (ELISA), restriction fragment length polymorphism (RFLP), denaturing gradient gel electrophoresis (DGGE), ** susceptibility is not restricted to the hosts mentioned in the table.
3. Isothermal Nucleic Acid Detection Methods
There is a need to develop techniques for the early identification of pathogens in farmed molluscs to allow for the decision-making process for controlling the infection, isolating, and treating the infected members, which can decrease the chances of mass outbreaks. Improved molecular technology for the detection of pathogen nucleic acid, such as polymerase chain reaction (PCR) and ISH, has been developed for a range of diseases affecting molluscs (Table 1). However, these require specialized laboratories and trained personnel [80,166,167,168]. In recent years there has been development of several isothermal nucleic acid amplification technologies which have the advantage of being simple to use, low-cost, field-deployable, and return rapid results [19,169,170,171,172]. This section outlines the isothermal amplification techniques (Figure 2) that have been applied to mollusc viral and parasitic disease diagnostics to fill the gap between quick diagnostics and accuracy.
3.1. Loop Mediated Isothermal Amplification (LAMP)
Loop-mediated isothermal amplification (LAMP) is one of the most widely used isothermal methods for detecting pathogens affecting molluscs (Table 2). The first LAMP assay was developed by Notomi et al. in 2000 to detect the hepatitis B surface antigen (HBs) region of the human hepatitis B virus. LAMP relies on an auto-cycling strand displacement mechanism for DNA synthesis. In this method, four to six primers are designed to target six to eight template regions in the presence of an isothermal DNA polymerase (Bst) [173,174]. The four main primers include two outer primers, F3 and B3, which function similarly to the forward and reverse primers in PCR. Additionally, two inner primers, the forward inner primer (FIP) and backward inner primer (BIP), are designed to complement two distinct regions on the sense and antisense strands of the target sequence. The FIP is composed of F2 and F1c, which is complementary to F2c on the sense strand and F1 on the antisense strand, respectively. The BIP is formed by connecting B2 and B1c, complementary to B2c on the antisense strand and B1 on the sense strand, respectively (Figure A2). The reaction can start by FIP or BIP, although here in we will start with FIP for easier explanation. The reaction begins when F2 of the FIP hybridizes with its complementary region F2c on the target strand. The F3 primer binds to an external region of the target and elongates, displacing the newly formed strand produced by FIP. The displaced strand, now containing the F1c region, anneals to its complementary F1 region on the new strand, forming a loop structure at one end. This new strand serves as a template for BIP. The B3 outer primer then displaces the new product formed by BIP, resulting in a dumbbell-shaped structure. The loop region acts as a template for the forward loop primer (FLP) and backward loop primer (BLP), providing additional amplification starting points. FIP, BIP, FLP, and BLP continue to prime on the loop and dumbbell-shaped DNA strands, leading to the formation of a cauliflower-like structure where multiple amplification, elongation, and displacement events occur simultaneously [173]. LAMP typically targets DNA samples or RNA after converting it to complementary DNA (cDNA). LAMP does not need extensive sample preparation, and is more tolerant to inhibitors than PCR [173,175]. The LAMP steps occur at a constant incubation temperature of 60–65 °C for 15-65 minutes, which eliminates the need for complicated instruments like thermocyclers. Instead, the process can be easily performed using a water bath or heat block. LAMP products can be visualized using various methods, including turbidity, colorimetry, electrochemical detection, agarose gel electrophoresis (AGE), or real-time detection with fluorescence [176,177,178,179].
3.2. Recombinase Polymerase Amplification (RPA)
Recombinase polymerase amplification (RPA) is another successful isothermal nucleic acid amplification technique, first developed by Piepenburg et al. in 2006 [169]. This method utilizes cellular DNA proteins with functions in synthesis, repair, and recombination. RPA requires only two primers (forward and reverse), a recombinase protein derived from the T4 bacteriophage, a high molecular weight polyethylene glycol acting as a crowding agent, single-stranded DNA binding protein, strand-displacing DNA polymerase, nucleotides, and ATP. An optional probe can be included to increase the specificity of the assay [169,180,181,182]. The recombinase protein binds to one of the primers, forming a recombinase-primer complex in the presence of ATP and the crowding agent (Figure B2). This complex scans the double-stranded DNA until it locates a sequence complementary to the primer. The primer complex then invades the double-stranded DNA and hybridizes with the target region, leaving the complementary strand exposed and seeking to reunite with the original complementary strand. The single-strand binding protein stabilizes the exposed strand, preventing it from re-annealing. Once the complex disassembles, the DNA polymerase attaches to the 3’ end of the primer and elongates it. This cycle repeats until the end of the incubation time [169]. The optimal temperature for RPA amplification ranges between 37-42°C, and the process does not require a denaturation step, which differs from cPCR [180,181]. DNA or RNA amplification can be completed within 5-20 minutes, and the resulting amplicons can be observed by many tools including, AGE or lateral flow devices (LFD) [181,183]. RPA is currently commercialized by TwistDx, highlighting its ease of use by untrained personnel and its high inhibition tolerance, allowing for the rapid processing of various field samples within minutes [184].
3.3. Cross-Priming Isothermal Amplification (CPA)
Cross-priming isothermal amplification (CPA) is a recently developed isothermal amplification method used to detect pathogens affecting molluscs. CPA was developed by Ustar Biotechnologies Co., Ltd. and utilizes 5-8 primers and probes, with at least one of them being a cross-linked primer [185,186,187]. CPA relies on the use of at least one cross primer, which consists of two connected sequences, one of them is complementary to the template and the second is designed as a flanking sequence at the 5’ end that allows for strand displacement. This flanking sequence is complementary to a region on the newly amplified strand, enabling the formation of a hairpin structure that serves as a template for further amplification by other primers [187]. Reverse primers are designed to anneal to the antisense strand in tandem, providing a region for the nicked double-stranded DNA to elongate with the help of the isothermal strand-displacing DNA polymerase (Bst) [185]. The reaction is initiated when the forward cross primer anneals to the sense strand and extends. An outer forward primer then anneals to a region upstream of the cross primer, displacing the newly synthesized strand (Figure C2). This newly displaced strand, with the 5’ end attachment, serves as a template for the reverse primers. The displacement of the reverse-primed strands results in two different amplicons: a shorter one that lacks the cross-linkage complementary sequence, and a longer one that contains the complementary region and forms a hairpin structure. These latter structures continue to act as templates for other primers, producing more of the previously described short and long amplicons, and the amplification continues until the reaction concludes [185]. CPA can amplify both DNA and RNA samples at temperatures ranging from 60 to 68°C without the need for an initial denaturation step [185,188]. The average amplification time is 40-60 minutes, and the products can be visualized using turbidity, colorimetry, or AGE [180,186,189,190].
3.4. Multiple Displacement Amplification (MDA)
Multiple displacement amplification (MDA) is a widely recognized isothermal amplification technique developed by Dean et al. in 2002. Initially called multiply-primed rolling circle amplification, and was originally designed to amplify plasmids. The technique relies on the use of random or partially random hexamer primers for strand-displacement, and amplification facilitated by the phi29 DNA polymerase. This enzyme is particularly suited for MDA due to its high strand-displacement capabilities, allowing it to synthesis up to 70,000 nucleotides in a single reaction. Additionally, phi29 DNA polymerase boasts exceptional fidelity and exonuclease resistance under isothermal conditions [191,192]. One of MDA's most notable applications is whole genome amplification (WGA). In WGA-MDA, random hexamer primers anneal to multiple sites on the template DNA, and phi29 DNA polymerase extends these primers across the entire template (Figure D2). As new strands displace the original complementary strands, they themselves become templates for further amplification. This reaction produces large amounts of highly branched DNA [191]. Unlike other isothermal amplification techniques such as LAMP, RPA, and CPA, MDA requires an initial denaturation step at 94°C for several minutes, followed by incubation with the enzyme at 30°C for several hours. The reaction is terminated by deactivating the enzyme at 65°C [193,194]. The efficiency of the assay can be improved by adding exonuclease-resistant primers to the 3’ end of the template or by using crowding agents [191]. The success of amplification can be visualized using AGE [195]. WGA-MDA can amplify DNA and RNA samples and is especially useful for amplifying very small quantities of genetic material. It can generate approximately 20 µg of DNA from a single genomic DNA copy, which is invaluable for samples that cannot be enriched or cultured [19,192,196,197]. However, WGA-MDA is highly sensitive to contaminants because of its lack of specificity (random amplification) and must be performed under strictly sterile conditions [195,198].
4. Application of Isothermal Amplification of Viral Pathogens Infecting Molluscs
The use of isothermal amplification has seen a remarkable increase over the last decade, offering a valuable option for pathogen detection directly in the field. This technology enables faster decision-making and more effective infection control, significantly enhancing the ability to respond to outbreaks and manage health risks in farmed molluscs. In 2014, four LAMP primers were designed to target a sequence of the DNA polymerase gene which is specific to AbHV. The assay was performed at 63°C for an hour and could detect as low as 100 virus copies/µl in nerve tissues extracted from moribund abalone. The results could be observed by AGE as well as naked eye by using UV light to visualize the fluorescent dye [176]. Furthermore, a real-time RPA assay was also designed to detect AbHV, the assay was faster and more sensitive than the corresponding qPCR test [170]. Genomic DNA was extracted from the muscle tissue of infected H. diversicolor, and specific primers and a probe were designed to amplify ORF49 (also called ORF38) of AbHV. The AbHV-RPA assay could detect 100 copies/reaction in 20 minutes at 37°C, with no cross-reactivity with any of the closely related pathogens, the results could be observed in real-time as well as AGE technique [170]. Both isothermal techniques now need to be performed in-field conditions to fully validate their future use.
To safeguard abalone and scallops from pathogenic viral infections, various LAMP assays have been developed as rapid and sensitive diagnostic tools. A LAMP assay targeting the AVNV was optimized for detection in scallop kidney tissues. Using four LAMP primers and a water bath as the heat source, the assay demonstrated a detection sensitivity of approximately 1 fg of genomic DNA—twice as sensitive as cPCR. Amplification results were visually confirmed with GeneFinder™ dye after one hour of incubation [199]. Additionally, a LAMP assay targeting the ORF2 sequence of AbSV achieved a detection limit of just 10 copies of AbSV vectors within one hour at 60°C, with amplification visualized using SYBR Green I dye and AGE [200]. While these assays exhibit high sensitivity and specificity, incorporating non-invasive sampling methods would enhance their field deployability, and the inclusion of loop primers could further accelerate reaction kinetics, reducing amplification time and improving overall diagnostic efficiency.
Various isothermal diagnostic tools have been developed to enhance the efficiency of OsHV-1 detection. A LAMP assay targeting the ATPase subunit of the OsHV-1 DNA-packaging terminase gene, encoded by ORF109, demonstrated a detection limit as low as 20 copies. Using four primers, the assay successfully detected the virus within one hour at 60°C from oyster tissues. However, the detection dye had to be added post-amplification, posing a risk of cross-contamination [201]. Later, an attempt for in-field use without additional handling termed single tube LAMP assay was developed to detect ORF4 sequence of OsHV-1. One step reaction was performed by adding hydroxynaphthol blue (HNB) dye to the reaction mix to avoid the potential contamination from the aforementioned LAMP assay [202]. Although the duration of this assay wase reduced to 10 minutes instead of one hour by adding a helicase enzyme, further improvements are still needed to enhance its sensitivity, which is currently around 1,000 copies [202].
Furthermore, a real-time RPA assay was designed to target ORF95 of OsHV-1 in genomic DNA samples extracted from ark clams [203]. In 2020, the same primers and probe from this assay were utilized to develop an in-field isothermal detection method by combining RPA with electrochemical detection. This new assay took 20 minutes to amplify a minimum of 207 copies of OsHV-1 at room temperature, whereas the original RPA assay was capable of detecting as low as five copies within the same time frame [203,204]. A CPA assay was also designed to specifically amplify the variant SB strain of OsHV-1 which was associated with mass mortalities in blood clams broodstocks [205]. Primers were generated targeting the conserved sequence of the OsHV-1-SB strain. The reaction was performed for an hour at 63°C and could detect as low as 30 copies/µl of the positive control plasmid. The assay is suitable for farms and poorly equipped laboratories as the results can be observed after simple centrifugation or by using dye GeneFinderTM [171].
5. Application of Isothermal Amplification of Parasitic Pathogens Infecting Molluscs
Isothermal amplification, particularly Loop-mediated Isothermal Amplification (LAMP), has shown significant promise in the field of mollusc disease diagnostics, providing a means to overcome the limitations of traditional detection methods. For instance, Bonamia species, such as B. exitiosa and B. ostreae, are unculturable protozoa, which presents a challenge in their detection and diagnosis [19]. To address this, three LAMP assays have been developed and evaluated to detect B. exitiosa, B. ostreae and Bonamia genus members [206]. The assays were tested using portable real-time Geni devices and a real-time thermocycler. Each of the developed assays employed six LAMP primers individually targeting unique regions of Actin, Actin-1 and 18S specific to B. exitiosa, B. ostreae and Bonamia genus, respectively. The B. exitiosa and B. ostreae LAMP assays were species-specific with a limit of detection of 50 copies/µL in a 30 minute reaction time, which was only 10 fold less sensitive than the qPCR. Notably, the B. exitiosa LAMP assay did not cross react with any of the non-target samples, B. ostreae LAMP assay detected Mikrocytos veneroïdes after 30 minutes and therefore, was deemed negative. The generic assay detected as low as 10 copies/µL, but the amplification was unreliable as successful amplification occurred in only five out of 10 runs. Therefore, the limit of detection was set to be 50 copies/µL, which is similar to the corresponding qPCR assay. Furthermore, the generic LAMP assay exhibited cross-reactivity with samples highly infected with Haplosporidium costale after 26 minutes with similar melting temperature as the target [206]. This latent amplification of Haplosporidium costale necessities confirmatory testing in regions where both pathogens may be present.
Many innovative methodologies are being developed to overcome the challenges posed by low DNA concentrations in diagnostic applications, with the goal of enhancing detection sensitivity. One such promising approach is whole genome amplification (WGA) using the multiple displacement amplification (MDA) technique, which has demonstrated success as an enrichment method. This technique has been applied to increase the number of B. exitiosa genomic DNA copies extracted from the gills of various mollusc species. The amplification process was carried out using the Illustra GenomiPhi V2 Amplification Kit (GE Healthcare), which contains Phi29 DNA polymerase, renowned for its high processivity and strand-displacement activity. The amplification was conducted at an optimal temperature of 30°C for a duration of 90 minutes. After the WGA-MDA step, PCR amplification was performed to target the actin gene of B. exitiosa [19]. Although WGA-MDA is not a direct detection tool, it plays a crucial role in enhancing the genomic DNA quantity for subsequent diagnostic assays. This isothermal technique enables the amplification of B. exitiosa genomic material even when the initial DNA concentration is low, thereby improving diagnostic accuracy and reliability for pathogens that possess minimal genomic DNA quantities.
This assay demonstrated remarkable sensitivity, capable of detecting as low as 20 fg of the pathogen, which is 100 times more sensitive than cPCR. The detection process was completed within 60 minutes [207]. These results underscore the enhanced efficiency and sensitivity of LAMP compared to cPCR, highlighting its potential as a rapid and highly sensitive diagnostic tool for detecting low-abundance pathogens.
Additionally, two LAMP assays were designed to identify members of the Perkinsus genus by targeting the conserved internal transcribed spacer 2 (ITS-2) region [177,208]. In the first LAMP assay, gill samples from living clams and suspected infected oysters were amplified using six specific LAMP primers [177]. The limit of detection for this assay was determined using a recombinant plasmid, which established a detection threshold of 10 copies within 50 minutes. This assay successfully detected 56 out of 60 positive samples, demonstrating greater sensitivity compared to the gold standard detection method, the RFTM, which identified 52 samples. The amplification results could be visually observed through turbidity changes or ultraviolet (UV) fluorescence [177]. The second LAMP assay was applied to tissues of Cortez and Pacific oysters [208]. This assay exhibited a detection sensitivity as low as 3.6 ng of DNA within 60 minutes, with amplification visualized through AGE or by adding SYBR Safe dye to the amplified products [208]. These results highlight the increasing success and widespread application of isothermal amplification techniques, such as LAMP, for the detection of a broader range of pathogens. Furthermore, LAMP assays demonstrate superior performance compared to traditional methods, offering improved sensitivity, faster detection times, and more accessible visualization techniques.
Furthermore, P. beihaiensis could be detected using RPA, after 25 minutes of incubation at 37°C, the amplification can be detected using LFD. The RPA-LFD assay could detect as low as 26 copies of ITS region specific for P. beihaiensis [209]. The sensitivity of the assay was equal to the corresponding qPCR assay when tested on gill samples from oysters, C. hongkongensis [209]. Additionally, six primers of LAMP were designed specifically to target the P. olseni conserved area between 5.8S rRNA and ITS2. The assay was species-specific with sensitivity up to 100 fg plasmid, kelly color was observed in positive samples of oysters while no colour change was observed with non- P. olseni samples [172]. Another LAMP assay was developed to amplify the 5.8S rDNA ITS sequences of P. olseni. Four primers could amplify as low as 30 copies of recombinant plasmid in one hour at 64°C, and didn’t show any cross reactivity with other members of the genus Perkinsus [210]. Continuous research is being conducted to improve the sensitivity and specificity of isothermal methods, however, more work needs to focus on simplifying these assays so they can be commercialized and used by farmworkers for regular surveillance before pathogens spread.
Table 2.
The current isothermal assays used to detect pathogens affecting molluscs.
| Pathogen | Type | Target | Sample | Duration (minutes) |
Sensitivity# | In-field | Ref. |
|---|---|---|---|---|---|---|---|
| Virus | |||||||
| Abalone herpesvirus (AVG) | LAMP | DNA polymerase gene | Nerve tissues | 60 | 100 copies/µL | No | [176] |
| Abalone herpesvirus (AVG) | RPA | ORF38 | Muscle tissue | 20 | 100 copies | No | [170] |
| Acute Viral Necrobiotic Virus (AVNV) | LAMP | - | Tissues | 60 | 1 fg | No | [199] |
| Abalone shrivelling syndrome associated virus (AbSV) | LAMP | ORF2 | Water | 60 | 10 copies | No | [200] |
| OsHV-1 | LAMP | ORF 109 | Tissues except for gonad and adductor muscle | 60 | 20 copies | No | [201] |
| OsHV-1 | LAMP | ORF 4 | Tissues | 60 | 103 copies | No | [202] |
| OsHV-1 | RPA | ORF 95 | Tissues | 20 | 207 copies | No | [204] |
| OsHV-1 | RPA | ORF 95 | Tissues | 20 | 5 copies | No | [203] |
| OsHV-1- SB* | CPA | - | - | 60 | 30 copies /µL | No | [171] |
| Parasites | |||||||
| B. exitiosa | MDA-WGA | Actin | Gill tissues | 90 | - | No | [19] |
| B. exitiosa | LAMP | Actin | Gill tissues | 30 | 50 copies/µL | No | [206] |
| B. ostreae | LAMP | Actin-1 | Gill tissues | 30 | 50 copies/µL | No | [206] |
| Bonamia spp. | LAMP | 18S | Gill tissues | 30 | 50 copies/µL | No | [206] |
| Marteilia refringens | LAMP | - | 60 | 20 fg | No | [207] | |
| Perkinsus spp. | LAMP | Internal Transcribed spacer 2 (ITS-2) | Gills/ body tissues | 49.8 |
10 copies of plasmid DNA |
No | [177] |
| Perkinsus spp. | LAMP | ITS2 | Tissues | 30-60 | 3.6-36 ng | No | [208] |
| Perkinsus beihaiensis | RPA | ITS | Gills | 25 | 26 copies | No | [209] |
| Perkinsus olseni | LAMP | ITS 5.8S rDNA | - | 60 | 30 copies | No | [210] |
| Perkinsus olseni | LAMP | Between 5.8S and ITS 2 | - | - | 100 fg | No | [172] |
* Variant of the typical strain. # Sensitivity is calculated by reaction unless otherwise specified.
6. Future Improvements in Application of Isothermal Amplification
Despite the development of several isothermal amplification assays for pathogen diagnostics in molluscs, the majority have not yet been adopted for widespread field applications. Several areas of improvement could facilitate the broader and more rapid uptake of these assays in the future. A successful field-deployable assay should feature simple, easy-to-perform sampling and extraction methods, short incubation times, the ability to process multiple samples simultaneously, and a clear, interpretable output.
A key area for improvement is the adoption of non-invasive sampling methods. All organisms shed DNA into the environment, known as environmental DNA (eDNA), and the ability to detect pathogens from environmental samples, such as water, would significantly enhance field-based disease surveillance [211]. eDNA has been successfully used to detect P. marinus in water samples and Candidatus Xenohaliotis californiensis bacteria in fecal and seawater samples [125,212]. However, further investigation is required to determine the most effective filter membranes, such as Zetapor, gauze, nylon, low-density polyethylene (LDPE), and polyvinylidene difluoride (PVDF), for capturing and eluting eDNA, considering their varying nucleic acid adsorption capacities and the specific needs for detecting different pathogens [213].
While isothermal amplification methods have demonstrated high sensitivity and specificity, further improvements can be achieved by incorporating enrichment techniques to overcome the challenges posed by low pathogen concentrations in samples. One promising approach involves the use of magnetic beads coated with anionic polymers, which have been shown to effectively capture viral pathogens in various sample matrices [214,215,216,217]. This method demonstrated high capture efficiency for viral pathogens, such as Human influenza A virus and Human immunodeficiency virus type-1 (HIV-1), with efficiencies ranging from 74% to 100%. However, lower efficiencies (5%-35%) were observed for other viruses, such as Vaccinia virus and Human herpesvirus 8, when testing water samples [216].
Currently, animal tissue samples are typically used for diagnostics, with DNA extraction performed using commercial kits [19,170]. However, efficient and rapid extraction can also be achieved using simpler methods, such as adding alkaline polyethylene glycol and heating [218]. This method offers several advantages over other chemical extraction methods, including eliminating the need for neutralization before PCR, making it suitable for viral, parasitic, and bacterial pathogens. Another simple and cost effective lysis method such as boiling tissue, was shown to yield higher DNA concentrations than five commercial extraction kits when applied to various food types, including bacon and fish eggs [219].
Most of the developed assays currently require approximately one hour to produce results, although some can deliver results in as little as 20 minutes (Table 2). The long incubation times, however, limit the throughput of samples and delay decision-making during disease outbreaks. Several strategies can be employed to accelerate assay results. For example, incorporating loop primers in LAMP reactions has been shown to reduce incubation time by half, potentially shortening the assay duration, which traditionally uses only four primers, if the sequence allows for the addition of loop primers [202,220,221]. Betaine has also been tested to improve specificity and sensitivity in recombinase polymerase amplification (RPA) assays. A study on RPA for hepatitis B virus detection demonstrated that the addition of 0.8 M of betaine improved test specificity and reduced the assay duration, without the need for a purification step [222]. Further testing is necessary to identify the most effective reagents for field-based detection.
Using rapid assays with portable, affordable readout methods like lateral flow devices can enhance field deployability and increase sample processing capacity [223,224]. Another promising readout method involves an electrochemical biosensor coupled with isothermal RPA for detecting OsHV-1 [204]. Ongoing research is essential to refine and optimize these detection methods, ultimately qualifying them for point-of-care use. This will be crucial for more effective disease control and prevention in aquaculture settings.
7. Conclusions
Mollusc production has been recognized for its environmental benefits and its role in maintaining a balanced ecosystem [2,5]. These advantages have fueled global expansion in mollusc farming. However, this increased production has also led to a rise in disease outbreaks, which have had significant economic and socioeconomic impacts on mollusc-producing countries. Given that molluscs lack an adaptive immune system, traditional vaccine approaches are not applicable, and most diseases have no available cure [225]. Therefore, early detection is crucial to controlling the spread of infections. Most of the standard molecular detection methods mentioned in this review are not highly specific, are time-consuming, expensive, require trained personnel, and depend on a constant power supply. The absence of cell lines to isolate viral pathogens that affect molluscs, combined with the different pathogen-host immune mechanisms, emphasizes the need for rapid pathogen detection, especially for viruses, which spread quickly and cause severe damage [226]. Isothermal detection assays offer a cheaper, faster, simpler alternative that do not require multiple expensive devices. However, it is rare to find an assay that has been tested under field conditions. More research is needed to develop suitable, contaminant-tolerant extraction and end-product visualization methods that are fast and more appropriate for field use. The constraints of time, cost, and specificity demand a shift towards replacing current gold standard detection methods with more efficient, fast, and sensitive techniques, such as isothermal detection methods [169].
Author Contributions
Conceptualization, H.A., K.H. and T.B.; data curation, H.A.; writing—original draft preparation, H.A. and G.Z.; figures H.A. and A.K.; writing—review and editing, H.A., G.Z., A.K., D.A., J.A., K.H. and T.B.; supervision, T.B.; funding acquisition, T.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by Cooperative Research Centers Project (CRC-P) awarded to Geneworks and La Trobe University.
Institutional Review Board Statement
Not applicable.
Acknowledgments
Authors would like to thank members of the Beddoe lab for their valuable advice during the preparation of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Worldometer. [cited 2024 04/10/2022]; Available from: https://www.worldometers.info/world-population/.
- Carnegie, R.B., I. Arzul, and D. Bushek, Managing marine mollusc diseases in the context of regional and international commerce: policy issues and emerging concerns. Philos. Trans. R. Soc. B, 2016. 371(1689): p. 20150215. [CrossRef]
- FAO, The State of World Fisheries and Aquaculture 2018-Meeting the sustainable development goals, in Fisheries and Aquaculture Department, Food and Agriculture Organization of the United Nations, Rome. 2020.
- FAO, The State of World Fisheries and Aquaculture 2024. Blue Transformation in action, in The State of World Fisheries and Aquaculture (SOFIA). 2024, FAO ;: Rome, Italy ;.
- Benkendorff, K., Molluscan biological and chemical diversity: Secondary metabolites and medicinal resources produced by marine molluscs. Biol. Rev, 2010. 85(4): p. 757-775. [CrossRef]
- Thakur, S., S. Singh, and B. Pal, Superior adsorption removal of dye and high catalytic activity for transesterification reaction displayed by crystalline CaO nanocubes extracted from mollusc shells. FPT, 2021. 213: p. 106707. [CrossRef]
- Marín Aguilera, B., F. Iacono, and M. Gleba, Colouring the mediterranean: Production and consumption of purple-dyed textiles in pre-roman times. J. Mediterr. Archaeol., 2019. [CrossRef]
- Meng, X., et al., Mannan oligosaccharide increases the growth performance, immunity and resistance capability against Vibro Parahemolyticus in juvenile abalone Haliotis discus hannai Ino. Fish Shellfish Immunol., 2019. 94: p. 654-660. [CrossRef]
- Arzul, I., et al., Viruses infecting marine molluscs. J. Invertebr. Pathol., 2017. 147: p. 118-135. [CrossRef]
- Renault, T. and B. Novoa, Viruses infecting bivalve molluscs. Aquat. Living Resour., 2004. 17(4): p. 397-409. [CrossRef]
- Paillard, C., F. Le Roux, and J.J. Borrego, Bacterial disease in marine bivalves, a review of recent studies: trends and evolution. Aquat. Living Resour., 2004. 17(4): p. 477-498. [CrossRef]
- Berthe, F.C., et al., Marteiliosis in molluscs: a review. Aquat. Living Resour., 2004. 17(4): p. 433-448. [CrossRef]
- Villalba, A., et al., Perkinsosis in molluscs: a review. Aquat. Living Resour., 2004. 17(4): p. 411-432. [CrossRef]
- Gleason, F.H., et al., The roles of endolithic fungi in bioerosion and disease in marine ecosystems. I. General concepts. Mycology, 2017. 8(3): p. 205-215. [CrossRef]
- OIE. Notifiable Animal Diseases. [cited 2023 22nd November]; Available from: https://www.woah.org/en/what-we-do/animal-health-and-welfare/animal-diseases/?_tax_animal=aquatics%2Cmolluscs.
- Arzul, I., T. Renault, and C. Lipart, Experimental herpes-like viral infections in marine bivalves: demonstration of interspecies transmission. Dis. Aquat. Organ., 2001. 46(1): p. 1-6. [CrossRef]
- Arzul, I., et al., Evidence for interspecies transmission of oyster herpesvirus in marine bivalves. J. Gen. Virol., 2001. 82(4): p. 865-870. [CrossRef]
- Canier, L., et al., A new multiplex real-time PCR assay to improve the diagnosis of shellfish regulated parasites of the genus Marteilia and Bonamia. Prev. Vet. Med., 2020. 183: p. 105126. [CrossRef]
- Prado Alvarez, M., et al., Whole-genome amplification: a useful approach to characterize new genes in unculturable protozoan parasites such as Bonamia exitiosa. Parasitol., 2015. 142(12): p. 1523-1534. [CrossRef]
- Bai, C.M., et al., Susceptibility of two abalone species, Haliotis diversicolor supertexta and Haliotis discus hannai, to Haliotid herpesvirus 1 infection. J. Invertebr. Pathol., 2019. 160: p. 26-32. [CrossRef]
- Corbeil, S., Abalone Viral Ganglioneuritis. Pathogens, 2020. 9(9): p. 720. [CrossRef]
- Chen, I.-W., et al., Exploring the chronic mortality affecting abalones in taiwan: differentiation of abalone herpesvirus-associated acute infection from chronic mortality by pcr and in situ hybridization and histopathology. Taiwan Vet. J., 2016. 42(01): p. 1-9. [CrossRef]
- Mouton, A., An Epidemiological Study of Parasites Infecting The South African Abalone (Haliotis Midae) In Western Cape Aquaculture Facilities. 2011, University of Pretoria (South Africa).
- Pen Heng, C., et al., Herpes-like virus infection causing mortality of cultured abalone Haliotis diversicolor supertexta in Taiwan. Dis. Aquat. Organ., 2005. 65(1): p. 23-27. [CrossRef]
- Corbeil, S., et al., Abalone viral ganglioneuritis: Establishment and use of an experimental immersion challenge system for the study of abalone herpes virus infections in Australian abalone. Virus Res., 2012. 165(2): p. 207-213. [CrossRef]
- Corbeil, S., et al., Innate resistance of New Zealand paua to abalone viral ganglioneuritis. J. Invertebr. Pathol., 2017. 146: p. 31-35. [CrossRef]
- Wang, J., et al., Virus infection in cultured abalone, Haliotis diversicolor Reeve in Guangdong Province, China. J. Shellfish Res., 2004. 23(4): p. 1163-1169.
- Hooper, C., P. Hardy-Smith, and J. Handlinger, Ganglioneuritis causing high mortalities in farmed Australian abalone (Haliotis laevigata and Haliotis rubra). Aust. Vet. J., 2007. 85(5): p. 188-193. [CrossRef]
- Caraguel, C.G., et al., Diagnostic test accuracy when screening for Haliotid herpesvirus 1 (AbHV) in apparently healthy populations of Australian abalone Haliotis spp. Dis. Aquat. Organ., 2019. 136(2): p. 199-207. [CrossRef]
- Corbeil, S., et al., Development and validation of a TaqMan® PCR assay for the Australian abalone herpes-like virus. Dis. Aquat. Organ., 2010. 92(1): p. 1-10. [CrossRef]
- Chen, M.-H., et al., Evaluation of a bacteriophage-related chimeric marine virus associated with abalone mortality in Taiwan. Taiwan Vet. J., 2014. 40(02): p. 77-82. [CrossRef]
- Wang, J., Spring explosive epidemic disease of abalone in Dongshan district. Xiamen Univ. Nat. Sci., 1999. 38: p. 644-654.
- Handlinger, J., General pathology and diseases of abalone, in Aquaculture Pathophysiology, F.S.B. Kibenge, B. Baldisserotto, and R.S.-M. Chong, Editors. 2022, Academic Press. p. 405-447.
- Jiang, J.-Z., et al., Nested PCR detection of abalone shriveling syndrome-associated virus in China. J. Virol. Methods, 2012. 184(1-2): p. 21-26. [CrossRef]
- Jiang, J.Z., et al., Quantitative PCR detection for abalone shriveling syndrome-associated virus. J. Virol. Methods, 2012. 184(1): p. 15-20. [CrossRef]
- Zhuang, J., et al., A bacteriophage-related chimeric marine virus infecting abalone. PloS one, 2010. 5(11): p. e13850. [CrossRef]
- Yang, R., Molecular epidemiological investigation on abalone shriveling syndrome-associated virus and herpes-like virus and its related research. 2013, ShangHai, China: ShangHai Ocean University.
- Haixin, A., et al., Artificial infection of cultured scallop Chlamys farreri by pathogen from acute virus necrobiotic disease. JFSC, 2003. 10(5): p. 386-391.
- Tang, B., et al., Physiological and immune responses of zhikong scallop Chlamys farreri to the acute viral necrobiotic virus infection. Fish Shellfish Immunol., 2010. 29(1): p. 42-48. [CrossRef]
- Ai, H., et al., Artificial infection of cultured scallop Chlamys farreri by pathogen from acute virus necrobiotic disease. Zhongguo shui chan ke xue = Journal of fishery sciences of China, 2003. 10(5): p. 386-391.
- Chen, G., et al., A preliminary study of differentially expressed genes of the scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish Shellfish Immunol., 2013. 34(6): p. 1619-1627. [CrossRef]
- Ren, W., Detection methods, sequence of the complete genome of acute viral necrobiotic virus isolated from scallop Chlamys farreri. 2009, Ocean University China.
- Chongming, W., et al., Purification and ultrastructure of a spherical virus in cultured scallop Chlamys farreri. Shuichan xuebao, 2002. 26(2): p. 180-183.
- Fu, C., W. Song, and Y. Li, Monoclonal antibodies developed for detection of an epizootic virus associated with mass mortalities of cultured scallop Chlamys farreri. Dis. Aquat. Organ., 2005. 65(1): p. 17-22. [CrossRef]
- Yun, L., et al., Detection of Acute Virus Necrobiotic Disease Virus (AVND Virus) in Chlamys farreri Using ELISA Technique. Gaojishu Tongxun, 2003. 13(7): p. 90-92.
- Fuhrmann, M., et al., Ostreid herpesvirus disease, in Aquaculture Pathophysiology, F.S.B. Kibenge, B. Baldisserotto, and R.S.-M. Chong, Editors. 2022, Academic Press. p. 473-488.
- Burge, C.A., et al., The first detection of a novel OsHV-1 microvariant in San Diego, California, USA. J. Invertebr. Pathol., 2021. 184: p. 107636. [CrossRef]
- Roque, A., et al., First report of OsHV-1 microvar in Pacific oyster (Crassostrea gigas) cultured in Spain. Aquac., 2012. 324-325: p. 303-306. [CrossRef]
- Renault, T. and C. Lipart. Diagnosis of herpes-like virus infections in oysters using molecular techniques. in Aquaculture and water: fish culture, shellfish culture and water usage. 1998.
- Schikorski, D., et al., Experimental infection of Pacific oyster Crassostrea gigas spat by ostreid herpesvirus 1: demonstration of oyster spat susceptibility. Vet. Res., 2011. 42: p. 1-13. [CrossRef]
- Arzul, I., et al., Detection of oyster herpesvirus DNA and proteins in asymptomatic Crassostrea gigas adults. Virus Res., 2002. 84(1-2): p. 151-160. [CrossRef]
- Barbosa Solomieu, V., et al., Diagnosis of Ostreid herpesvirus 1 in fixed paraffin-embedded archival samples using PCR and in situ hybridisation. J. Virol. Methods, 2004. 119(2): p. 65-72. [CrossRef]
- Lipart, C. and T. Renault, Herpes-like virus detection in infected Crassostrea gigas spat using DIG-labelled probes. J. Virol. Methods, 2002. 101(1-2): p. 1-10. [CrossRef]
- Pepin, J.F., A. Riou, and T. Renault, Rapid and sensitive detection of ostreid herpesvirus 1 in oyster samples by real-time PCR. J. Virol. Methods, 2008. 149(2): p. 269-276. [CrossRef]
- Burreson, E.M. and S.E. Ford, A review of recent information on the Haplosporidia, with special reference to Haplosporidium nelsoni (MSX disease). Aquat. Living Resour., 2004. 17(4): p. 499-517. [CrossRef]
- Comps, M. and Y. Pichot, Fine spore structure of a haplosporidan parasitizing Crassostrea gigas: taxonomic implications. Dis. Aquat. Org, 1991. 11: p. 73-77.
- Andrews, J.D. Oyster mortality studies in Virginia IV. MSX in James River public seed beds. in Proceedings of the National Shellfisheries Association. 1964.
- Ford, S.E. and H.H. Haskin, History and epizootiology of Haplosporidium nelsoni (MSX), an oyster pathogen in Delaware Bay, 1957–1980. J. Invertebr. Pathol., 1982. 40(1): p. 118-141. [CrossRef]
- Ford, S.E., et al., Investigating the life cycle of Haplosporidium nelsoni (MSX): A review. J. Shellfish Res., 2018. 37(4): p. 679-693. [CrossRef]
- Barber, B.J., S.E. Ford, and D. Littlewood, A physiological comparison of resistant and susceptible oysters Crassostrea virginica (Gmelin) exposed to the endoparasite Haplosporidium nelsoni (Haskin, Stauber & Mackin). J. Exp. Mar. Bio. Ecol., 1991. 146(1): p. 101-112. [CrossRef]
- Stokes, N.A. and E.M. Burreson, Differential diagnosis of mixed Haplosporidium costale and Haplosporidium nelsoni infections in the eastern oyster, Crassostrea virginica, using DNA probes. J. Shellfish Res., 2001. 20(1): p. 207.
- Arzul, I. and R.B. Carnegie, New perspective on the haplosporidian parasites of molluscs. J. Invertebr. Pathol., 2015. 131: p. 32-42. [CrossRef]
- Penna, M.S., M. Khan, and R.A. French, Development of a multiplex PCR for the detection of Haplosporidium nelsoni, Haplosporidium costale andPerkinsus marinus in the eastern oyster (Crassostrea virginica, Gmelin, 1971). MCP, 2001. 15(6): p. 385-390. [CrossRef]
- Carnegie, R.B., et al., Bonamia perspora n. sp.(Haplosporidia), a parasite of the oyster Ostreola equestris, is the first Bonamia species known to produce spores. J. Eukaryot. Microbiol., 2006. 53(4): p. 232-245. [CrossRef]
- Dinamani, P., M. Hine, and J. Jones, Occurrence and characteristics of the haemocyte parasite Bonamia ostreae in the New Zealand dredge oyster Tiostrea lutaria. Dis. Aquat. Organ., 1987. 3: p. 37-44.
- Culloty, S.C. and M.F. Mulcahy, Bonamia ostreae in the native oyster Ostrea edulis. Fish. Bull. Mar. Inst. Dublin, 2007.
- European Commission, D.-G.f.H.a.F.S., Commission implementing regulations (EU) 2018/1882 in Official Journal of the European Union. 2022.
- Bower, S.M., S.E. McGladdery, and I.M. Price, Synopsis of infectious diseases and parasites of commercially exploited shellfish. Annu. Rev. Fish Dis., 1994. 4: p. 1-199. [CrossRef]
- Jorgensen, L.v.G., et al., A non-lethal method for detection of Bonamia ostreae in flat oyster (Ostrea edulis) using environmental DNA. Sci. Rep., 2020. 10(1): p. 16143. [CrossRef]
- Ramilo, A., et al., Species-specific diagnostic assays for Bonamia ostreae and B. exitiosa in European flat oyster Ostrea edulis: conventional, real-time and multiplex PCR. Dis. Aquat. Organ., 2013. 104(2): p. 149-161. [CrossRef]
- Pichot, Y., et al., Recherches sur Bonamia ostreae gen. n., sp. n., parasite nouveau de l'huître plate Ostrea edulis L. Rev. Trav. Off. Pech. Marit., 1979. 43(1): p. 131-140.
- Van Banning, P., The life cycle of the oyster pathogen Bonamia ostreae with a presumptive phase in the ovarian tissue of the European flat oyster, Ostrea edulis. Aquac., 1990. 84(2): p. 189-192. [CrossRef]
- Abollo, E., et al., First detection of the protozoan parasite Bonamia exitiosa (Haplosporidia) infecting flat oyster Ostrea edulis grown in European waters. Aquaculture, 2008. 274(2): p. 201-207. [CrossRef]
- Lane, H.S., Studies on Bonamia parasites (Haplosporidia) in the New Zealand flat oyster Ostrea chilensis. 2018, University of Otago.
- Hine, P., et al., Ultrastructure of Mikrocytos mackini, the cause of Denman Island disease in oysters Crassostrea spp. and Ostrea spp. in British Columbia, Canada. Dis. Aquat. Organ., 2001. 45(3): p. 215-227. [CrossRef]
- Culloty, S.C., M.A. Cronin, and M.F. Mulcahy, Potential resistance of a number of populations of the oyster Ostrea edulis to the parasite Bonamia ostreae. Aquac., 2004. 237(1): p. 41-58. [CrossRef]
- Van Banning, P., Observations on bonamiasis in the stock of the European flat oyster, Ostrea edulis, in the Netherlands, with special reference to the recent developments in Lake Grevelingen. Aquac., 1991. 93(3): p. 205-211. [CrossRef]
- Carnegie, R.B., et al., Development of a PCR assay for detection of the oyster pathogen Bonamia ostreae and support for its inclusion in the Haplosporidia. Dis. Aquat. Organ., 2000. 42(3): p. 199-206. [CrossRef]
- Cochennec, N., et al., Detection of Bonamia ostreae Based on Small Subunit Ribosomal Probe. J. Invertebr. Pathol., 2000. 76(1): p. 26-32. [CrossRef]
- Corbeil, S., et al., Development of a TaqMan PCR assay for the detection of Bonamia species. Dis. Aquat. Organ., 2006. 71(1): p. 75-80. [CrossRef]
- Hill, K.M., et al., Observation of a Bonamia sp. infecting the oyster Ostrea stentina in Tunisia, and a consideration of its phylogenetic affinities. J. Invertebr. Pathol., 2010. 103(3): p. 179-185. [CrossRef]
- Carrasco, N., et al., Bonamia exitiosa (Haplosporidia) observed infecting the European flat oyster Ostrea edulis cultured on the Spanish Mediterranean coast. J. Invertebr. Pathol., 2012. 110(3): p. 307-313. [CrossRef]
- Hine, P., Severe apicomplexan infection in the oyster Ostrea chilensis: a possible predisposing factor in bonamiosis. Dis. Aquat. Organ., 2002. 51(1): p. 49-60. [CrossRef]
- Calvo, L.M.R., G.W. Calvo, and E.M. Burreson, Dual disease resistance in a selectively bred eastern oyster, Crassostrea virginica, strain tested in Chesapeake Bay. Aquac., 2003. 220(1-4): p. 69-87. [CrossRef]
- Comps, M., Marteilia lengehi n. sp., parasite de l'huître Crassostrea cucullata Born. Rev. Trav. Off. Pech. Marit., 1976. 40(2): p. 347-349.
- Perkins, F.O. and P.H. Wolf, Fine structure of Marteilia sydneyi sp. n.: haplosporidan pathogen of Australian oysters. J. Parasitol., 1976: p. 528-538. [CrossRef]
- Villalba, A., et al., Cockle Cerastoderma edule fishery collapse in the Ría de Arousa (Galicia, NW Spain) associated with the protistan parasite Marteilia cochillia. Dis. Aquat. Organ., 2014. 109(1): p. 55-80. [CrossRef]
- Norton, J., F. Perkins, and E. Ledua, Marteilia-like infection in a giant clam, Tridacna maxima, in Fiji. J. Invertebr. Pathol., 1993. [CrossRef]
- Carrasco, N., T. Green, and N. Itoh, Marteilia spp. parasites in bivalves: A revision of recent studies. J. Invertebr. Pathol., 2015. 131: p. 43-57. [CrossRef]
- Le Roux, F., et al., DNA probes as potential tools for the detection of Marteilia refringens. Mar. Biotechnol., 1999. 1(6): p. 588-597. [CrossRef]
- Lopez Flores, I., et al., The molecular diagnosis of Marteilia refringens and differentiation between Marteilia strains infecting oysters and mussels based on the rDNA IGS sequence. Parasitol., 2004. 129(4): p. 411-419. [CrossRef]
- Birch, G., C. Apostolatos, and S. Taylor, A remarkable recovery in the Sydney rock oyster (Saccostrea glomerata) population in a highly urbanised estuary (Sydney estuary, Australia). J. Coast. Res., 2013. 29(5): p. 1009-1015. [CrossRef]
- Grizel, H., Etude des récentes épizooties de l'huître plate Ostrea edulis Linné et de leur impact sur l'ostréiculture bretonne. 1985, Universite des sciences et techniques du Languedoc.
- Le Roux, F., et al., Molecular evidence for the existence of two species of Marteilia in Europe. J. Eukaryot. Microbiol., 2001. 48(4): p. 449-454. [CrossRef]
- Audemard, C., et al., Needle in a haystack: involvement of the copepod Paracartia grani in the life-cycle of the oyster pathogen Marteilia refringens. Parasitol., 2002. 124(3): p. 315-323. [CrossRef]
- Villalba, A., et al., Marteiliasis affecting cultured mussels Mytilus galloprovincialis of Galicia (NW Spain). I. Etiology, phases of the infection, and temporal and spatial variability in prevalence. Dis. Aquat. Organ., 1993. [CrossRef]
- Wetchateng, T., et al., Withering syndrome in the abalone Haliotis diversicolor supertexta. Dis. Aquat. Organ., 2010. 90(1): p. 69-76. [CrossRef]
- Grizel, H., et al., Recherche sur l'agent de la maladie de la glande digestive de Ostrea edulis Linné. Sci. Pech. , 1974(240): p. 7-30.
- Grizel, H., et al., Etude d'un parasite de la glande digestive observé au cours de l'épizootie actuelle de l'huître plate. C. R. Acad. Sci., 1974. 279: p. 783-785.
- Thébault, A., et al., Validation of in situ hybridisation and histology assays for the detection of the oyster parasite Marteilia refringens. Dis. Aquat. Organ., 2005. 65(1): p. 9-16. [CrossRef]
- Chun, S.K., Preliminary studies on the sporozoan parasites in oysters on the southern coast of Korea. Bull. Korean Fish. Soc., 1972. 5: p. 76-82.
- Comps, M. and I. Desportes, Etude ultrastructurale des Marteilioides chungmuensis NG, N. SP. parasite des ovocytes de l'huître Crassostrea gigas Th. Protist., 1986. 22(3): p. 279-285.
- Itoh, N., et al., DNA probes for detection of Marteilioides chungmuensis from the ovary of Pacific oyster Crassostrea gigas. Fish Pathol., 2003. 38(4): p. 163-169. [CrossRef]
- Ngo, T.T., F.C. Berthe, and K.-S. Choi, Prevalence and infection intensity of the ovarian parasite Marteilioides chungmuensis during an annual reproductive cycle of the oyster Crassostrea gigas. Dis. Aquat. Organ., 2003. 56(3): p. 259-267. [CrossRef]
- Anderson, T. and R. Lester, Sporulation of Marteilioides branchialis n. sp.(Paramyxea) in the Sydney rock oyster, Saccostrea commercialis: an electron microscope study. The Journal of protozoology, 1992. 39(4): p. 502-508. [CrossRef]
- Choi, H.J., et al., A study of diagnostic methods for Marteilioides chungmuensis infections in the Pacific oyster Crassostrea gigas. "J. Invertebr. Pathol., 2012. 111(1): p. 27-32. [CrossRef]
- Burki, F., et al., Phylogenomics of the Intracellular Parasite Mikrocytos mackini Reveals Evidence for a Mitosome in Rhizaria. Curr. Biol., 2013. 23(16): p. 1541-1547. [CrossRef]
- Carnegie, R.B., B.J. Barber, and D.L. Distel, Detection of the oyster parasite Bonamia ostreae by fluorescent in situ hybridization. Dis. Aquat. Organ., 2003. 55(3): p. 247-252. [CrossRef]
- Polinski, M., et al., Molecular detection of Mikrocytos mackini in Pacific oysters using quantitative PCR. Mol Biochem Parasitol., 2015. 200(1-2): p. 19-24. [CrossRef]
- Auzoux-Bordenave, S., et al., In vitro sporulation of the clam pathogen Perkinsus atlanticus (Apicomplexa, Perkinsea) under various environmental conditions. Oceanogr. Lit. Rev., 1996. 9(43): p. 926.
- Dantas Neto, M.P., et al., First record of Perkinsus chesapeaki infecting Crassostrea rhizophorae in South America. J. Invertebr. Pathol., 2016. 141: p. 53-56. [CrossRef]
- Park, K.I. and K.S. Choi, Application of enzyme-linked immunosorbent assay for studying of reproduction in the Manila clam Ruditapes philippinarum (Mollusca: Bivalvia): I. Quantifying eggs. Aquac., 2004. 241(1-4): p. 667-687. [CrossRef]
- Park, K.-I., et al., Pathology survey of the short-neck clam Ruditapes philippinarum occurring on sandy tidal flats along the coast of Ariake Bay, Kyushu, Japan. J. Invertebr. Pathol., 2008. 99(2): p. 212-219. [CrossRef]
- Neto, M.P.D., et al., First record of Perkinsus chesapeaki infecting Crassostrea rhizophorae in South America. J. Invertebr. Pathol., 2016. 141: p. 53-56. [CrossRef]
- Hanrio, E., et al., Immunoassays and diagnostic antibodies for Perkinsus spp. pathogens of marine molluscs. Dis. Aquat. Organ., 2021. 147: p. 13-23. [CrossRef]
- Dungan, C.F. and K.S. Reece, 5.2. 1 Perkinsus spp. Infections of Marine Molluscs (2020). AFS-FHS, 2020.
- Valencia, J., et al., New data on Perkinsus mediterraneus in the Balearic Archipelago: locations and affected species. Dis. Aquat. Organ., 2014. 112(1): p. 69-82. [CrossRef]
- Andrews, J.D., Epizootiology of the disease caused by the oyster pathogen Perkinsus marinus and its effects on the oyster industry. American Fisheries Society, 1988. 18.
- Cook, T., et al., The Relationship Between Increasing Sea-surface Temperature and the Northward Spread ofPerkinsus marinus (Dermo) Disease Epizootics in Oysters. Estuar. Coast. Shelf Sci., 1998. 46(4): p. 587-597. [CrossRef]
- Pires, D., et al., Histopathologic Lesions in Bivalve Mollusks Found in Portugal: Etiology and Risk Factors. J. mar. sci. eng., 2022. 10(2): p. 133. [CrossRef]
- Casas, S.M., A. Villalba, and K.S. Reece, Study of perkinsosis in the carpet shell clam Tapes decussatus in Galicia (NW Spain). I. Identification of the aetiological agent and in vitro modulation of zoosporulation by temperature and salinity. Dis. Aquat. Organ., 2002. 50(1): p. 51-65. [CrossRef]
- Gauthier, J., C. Miller, and A.E. Wilbur, TaqMan® MGB real-time PCR approach to quantification of Perkinsus marinus and Perkinsus spp. in oysters. J. Shellfish Res., 2006. 25(2): p. 619-624. [CrossRef]
- Lenaers, G., et al., Dinoflagellates in evolution. A molecular phylogenetic analysis of large subunit ribosomal RNA. J. Mol. Evol., 1989. 29(1): p. 40-51. [CrossRef]
- Moss, J.A., et al., Description of Perkinsus beihaiensis n. sp., a new Perkinsus sp. parasite in oysters of southern China. J. Eukaryot. Microbiol., 2008. 55(2): p. 117-130. [CrossRef]
- Audemard, C., K.S. Reece, and E.M. Burreson, Real-time PCR for detection and quantification of the protistan parasite Perkinsus marinus in environmental waters. Appl. Environ. Microbiol., 2004. 70(11): p. 6611-6618. [CrossRef]
- Elston, R.A., et al., Perkiasus sp infection risk for manila clams, Venerupis philippinarum (A. Adams and Reeve, 1850) on the Pacific coast of North and Central America. J. Shellfish Res., 2004. 23(1): p. 101.
- Ramilo, A., et al., Update of information on perkinsosis in NW Mediterranean coast: Identification of Perkinsus spp.(Protista) in new locations and hosts. J. Invertebr. Pathol., 2015. 125: p. 37-41. [CrossRef]
- Reece, K.S., C.F. Dungan, and E.M. Burreson, Molecular epizootiology of Perkinsus marinus and P. chesapeaki infections among wild oysters and clams in Chesapeake Bay, USA. Dis. Aquat. Organ., 2008. 82(3): p. 237-248. [CrossRef]
- COSS, C.A., et al., Description of Perkinsus andrewsi n. sp. isolated from the Baltic clam (Macoma balthica) by characterization of the ribosomal RNA locus, and development of a species-specific PCR-based diagnostic assay. J. Eukaryot. Microbiol., 2001. 48(1): p. 52-61. [CrossRef]
- Kang, H.-S., et al., Survey on Perkinsus species in Manila clam Ruditapes philippinarum in Korean waters using species-specific PCR. Fish Pathol., 2017. 52(4): p. 202-205. [CrossRef]
- Elandalloussi, L.M., et al., Development of a PCR-ELISA assay for diagnosis of Perkinsus marinus and Perkinsus atlanticus infections in bivalve molluscs. MCP, 2004. 18(2): p. 89-96. [CrossRef]
- Moss, J.A., Characterization of exotic pathogens associated with the suminoe oyster, Crassostrea ariakensis. 2007.
- Chen, M.H., et al., Development of a polymerase chain reaction for the detection of abalone herpesvirus infection based on the DNA polymerase gene. J. Virol. Methods, 2012. 185(1): p. 1-6. [CrossRef]
- Fegan, M., et al. Development of an in situ hybridisation assay for the detection and identification of the abalone herpes-like virus. in 4th FRDC Aquatic Animal Health Subprogram Scientific Conference, Cairns. 2009.
- Ren, W., et al., Development and application of a FQ-PCR assay for detection of Chlamys farreri acute viral necrobiotic virus. JFSC, 2009. 16(4): p. 564-571.
- Xing, J., T. Lin, and W. Zhan, Variations of enzyme activities in the haemocytes of scallop Chlamys farreri after infection with the acute virus necrobiotic virus (AVNV). Fish Shellfish Immunol., 2008. 25(6): p. 847-852. [CrossRef]
- Narcisi, V., et al., Detection of Bonamia ostreae and B. exitiosa (Haplosporidia) in Ostrea edulis from the Adriatic Sea (Italy). Dis. Aquat. Organ., 2010. 89(1): p. 79-85. [CrossRef]
- Carnegie, R.B. and N. Cochennec-Laureau, Microcell parasites of oysters: recent insights and future trends. Aquat. Living Resour., 2004. 17(4): p. 519-528. [CrossRef]
- Ramilo, A., A. Villalba, and E. Abollo, Species-specific oligonucleotide probe for detection of Bonamia exitiosa (Haplosporidia) using in situ hybridisation assay. Dis. Aquat. Organ., 2014. 110(1-2): p. 81-91. [CrossRef]
- Marty, G.D., et al., Histopathology and a real-time PCR assay for detection of Bonamia ostreae in Ostrea edulis cultured in western Canada. Aquac., 2006. 261(1): p. 33-42. [CrossRef]
- Robert, M., et al., Molecular detection and quantification of the protozoan Bonamia ostreae in the flat oyster, Ostrea edulis. MCP, 2009. 23(6): p. 264-271. [CrossRef]
- Engelsma, M.Y., et al., Epidemiology of Bonamia ostreae infecting european flat oysters ostrea edulis from Lake Grevelingen, The Netherlands. Mar. Ecol. Prog. Ser., 2010. 409: p. 131-142. [CrossRef]
- Xie, Z., et al., A duplex quantitative real-time PCR assay for the detection of Haplosporidium and Perkinsus species in shellfish. Parasitol. Res., 2013. 112(4): p. 1597-1606. [CrossRef]
- Arzul, I., et al., First characterization of the parasite Haplosporidium costale in France and development of a real-time PCR assay for its rapid detection in the Pacific oyster, Crassostrea gigas. Transbound. Emerg. Dis., 2022. 69(5): p. e2041-e2058. [CrossRef]
- Lopez Nuñez, R., et al., Detection of Haplosporidium pinnae from Pinna nobilis Faeces. J. mar. sci. eng., 2022. 10(2): p. 276. [CrossRef]
- Carella, F., et al., In the wake of the ongoing mass mortality events: Co-occurrence of Mycobacterium, Haplosporidium and other pathogens in Pinna nobilis collected in Italy and Spain (Mediterranean Sea). Front. mar. sci., 2020. 7: p. 48. [CrossRef]
- Stokes, N.A., Burreson, Eugene M., A sensitive and specific DNA probe for the oyster pathogen Haplosporidium nelsoni. J. Eukaryot. Microbiol., 1995. 42(4): p. 350-357. [CrossRef]
- Stokes, N.A., M.E. Siddall, and E.M. Burreson, Detection of Haplosporidium nelsoni (Haplosporidia: Haplosporidiidae) in oysters by PCR amplification. Dis. Aquat. Organ., 1995. 23(2): p. 145-152. [CrossRef]
- Day, J.M., D.E. Franklin, and B.L. Brown, Use of Competitive PCR to Detect and Quantify Haplosporidium nelsoni Infection (MSX disease) in the Eastern Oyster (Crassostrea virginica). Mar Biotechnol (NY), 2000. 2(5): p. 456-465. [CrossRef]
- Carrasco, N., et al., Molecular characterization of the Marteilia parasite infecting the common edible cockle Cerastoderma edule in the Spanish Mediterranean coast: a new Marteilia species affecting bivalves in Europe? Aquac., 2012. 324: p. 20-26. [CrossRef]
- Pernas, M., et al., Molecular methods for the diagnosis of Marteilia refringens. B EUR ASSOC FISH PAT., 2001. 21(5): p. 200-208.
- Carrasco, N., et al., Application of a competitive real time PCR for detection of Marteilia refringens genotype “O” and “M” in two geographical locations: The Ebro Delta, Spain and the Rhine-Meuse Delta, the Netherlands. J. Invertebr. Pathol., 2017. 149: p. 51-55. [CrossRef]
- Berthe, F., et al., The existence of the phylum Paramyxea Desportes and Perkins, 1990 is validated by the phylogenetic analysis of the Marteilia refringens small subunit ribosomal RNA. J. Euk. Microbiol, 2000. 47: p. 288-293.
- Anderson, T., R. Adlard, and R. Lester, Molecular diagnosis of Marteilia sydneyi (Paramyxea) in Sydney rock oysters, Saccostrea commercialis (Angas). Journal of Fish Diseases, 1995. 18(6): p. 507-510. [CrossRef]
- Kleeman, S., et al., Specificity of PCR and in situ hybridization assays designed for detection of Marteilia sydneyi and M. refringens. Parasitology, 2002. 125(2): p. 131-141. [CrossRef]
- Yanin, L., et al., Molecular and histological identification of Marteilioides infection in Suminoe Oyster Crassostrea ariakensis, Manila Clam Ruditapes philippinarum and Pacific Oyster Crassostrea gigas on the south coast of Korea. J. Invertebr. Pathol., 2013. 114(3): p. 277-284. [CrossRef]
- Itoh, N., et al., An ovarian infection in the Iwagaki oyster, Crassostrea nippona, with the protozoan parasite Marteilioides chungmuensis. J. Fish Dis., 2004. 27(5): p. 311-314.
- Carnegie, R.B., et al., Molecular detection of the oyster parasite Mikrocytos mackini, and a preliminary phylogenetic analysis. Dis. Aquat. Organ., 2003. 54(3): p. 219-227. [CrossRef]
- Meyer, G.R., S.M. Bower, and R.B. Carnegie, Sensitivity of a digoxigenin-labelled DNA probe in detecting Mikrocytos mackini, causative agent of Denman Island disease (mikrocytosis), in oysters. J. Invertebr. Pathol., 2005. 88(2): p. 89-94. [CrossRef]
- Marsh, A.G., J.D. Gauthier, and G.R. Vasta, A semiquantitative PCR assay for assessing Perkinsus marinus infections in the eastern oyster, Crassostrea virginica. J. Parasitol., 1995: p. 577-583.
- Ramilo, A., et al., Perkinsus olseni and P. chesapeaki detected in a survey of perkinsosis of various clam species in Galicia (NW Spain) using PCR–DGGE as a screening tool. J. Invertebr. Pathol., 2016. 133: p. 50-58. [CrossRef]
- De la Herrán, R., et al., Molecular characterization of the ribosomal RNA gene region of Perkinsus atlanticus: its use in phylogenetic analysis and as a target for a molecular diagnosis. Parasitol., 2000. 120(4): p. 345-353. [CrossRef]
- Moss, J.A., E.M. Burreson, and K.S. Reece, Advanced Perkinsus marinus infections in Crassostrea ariakensis maintained under laboratory conditions. J. Shellfish Res., 2006. 25(1): p. 65-72. [CrossRef]
- Abollo, E., et al., Differential diagnosis of Perkinsus species by polymerase chain reaction-restriction fragment length polymorphism assay. Molecular and Cellular Probes, 2006. 20(6): p. 323-329. [CrossRef]
- Reece, K.S., et al., A novel monoclonal Perkinsus chesapeaki in vitro isolate from an Australian cockle, Anadara trapezia. J. Invertebr. Pathol., 2017. 148: p. 86-93. [CrossRef]
- Kim, H.J., et al., Mass mortality in Korean bay scallop (Argopecten irradians) associated with Ostreid Herpesvirus-1 μVar. Transbound. Emerg. Dis., 2019. 66(4): p. 1442-1448. [CrossRef]
- Roque, A., et al., Detection and identification of tdh-and trh-positive Vibrio parahaemolyticus strains from four species of cultured bivalve molluscs on the Spanish Mediterranean Coast. Appl. Environ. Microbiol., 2009. 75(23): p. 7574-7577. [CrossRef]
- Schikorski, D., et al., Development of TaqMan real-time PCR assays for monitoring Vibrio harveyi infection and a plasmid harbored by virulent strains in European abalone Haliotis tuberculata aquaculture. Aquac., 2013. 392-395: p. 106-112. [CrossRef]
- Piepenburg, O., et al., DNA detection using recombination proteins. PLoS Biol., 2006. 4(7): p. e204. [CrossRef]
- Gao, F., et al., Real-time isothermal detection of Abalone herpes-like virus and red-spotted grouper nervous necrosis virus using recombinase polymerase amplification. J. Virol. Methods, 2018. 251: p. 92-98. [CrossRef]
- Zhao, X., et al., Establishment and application of a cross priming isothermal amplification technique for detection of SB strain of Ostreid herpesvirus-1. J. Fish. China. , 2015. 39(4): p. 580-588.
- Pang, Y., et al., Establishment and application of visual LAMP assay on Perkinsus olseni. Southwest China Journal of Agricultural Sciences, 2012. 25(1): p. 302-305.
- Notomi, T., et al., Loop-mediated isothermal amplification of DNA. Nucleic acids res. , 2000. 28(12): p. e63-e63. [CrossRef]
- Fakruddin, M., Loop mediated isothermal amplification (LAMP)–an alternative to polymerase chain reaction (PCR). Bangladesh Res. Publ. J, 2011. 5(4).
- Tao, Y., et al., High-performance detection of Mycobacterium bovis in milk using digital LAMP. Food chem., 2020. 327: p. 126945. [CrossRef]
- Chen, M.H., et al., The development of a loop-mediated isothermal amplification assay for rapid and sensitive detection of abalone herpesvirus DNA. J. Virol. Methods, 2014. 196: p. 199-203. [CrossRef]
- Feng, C., et al., Development of a loop-mediated isothermal amplification method for detection of Perkinsus spp. in mollusks. Dis. Aquat. Organ., 2013. 104(2): p. 141-148. [CrossRef]
- Wang, D., et al., Development of a real-time loop-mediated isothermal amplification (LAMP) assay and visual LAMP assay for detection of African swine fever virus (ASFV). J. Virol. Methods, 2020. 276: p. 113775. [CrossRef]
- Biswas, G. and M. Sakai, Loop-mediated isothermal amplification (LAMP) assays for detection and identification of aquaculture pathogens: current state and perspectives. AMBB, 2014. 98(7): p. 2881-2895. [CrossRef]
- Zhong, J. and X. Zhao, Isothermal Amplification Technologies for the Detection of Foodborne Pathogens. Food Anal. Methods, 2018. 11(6): p. 1543-1560. [CrossRef]
- Daher, R.K., et al., Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem., 2016. 62(7): p. 947-958. [CrossRef]
- Lobato, I.M. and C.K. O'Sullivan, Recombinase polymerase amplification: Basics, applications and recent advances. TrAC, Trends Anal. Chem., 2018. 98: p. 19-35. [CrossRef]
- Dai, T., et al., Comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, conventional PCR, and leaf-disc baiting methods for detection of Phytophthora sojae. Front. microbiol., 2019. 10: p. 1884. [CrossRef]
- TwistDx. TwistDx. [cited 2024 02/09]; Available from: https://www.twistdx.co.uk/rpa/.
- Xu, G., et al., Cross priming amplification: mechanism and optimization for isothermal DNA amplification. Sci Rep, 2012. 2: p. 246. [CrossRef]
- Kumar, Y., Isothermal amplification-based methods for assessment of microbiological safety and authenticity of meat and meat products. Food Control, 2021. 121: p. 107679. [CrossRef]
- Fang, R., et al., Cross-Priming Amplification for Rapid Detection of Mycobacterium tuberculosis in Sputum Specimens. J. Clin. Microbiol., 2009. 47(3): p. 845-847. [CrossRef]
- Bai, Z., et al., Isothermal cross-priming amplification implementation study. Lett. Appl. Microbiol, 2015. 60(3): p. 205-209. [CrossRef]
- Xu, Z., et al., Rapid Detection of Food-Borne Escherichia coli O157:H7 with Visual Inspection by Crossing Priming Amplification (CPA). Food Anal. Methods, 2020. 13(2): p. 474-481. [CrossRef]
- Wang, Y., et al., Rapid and sensitive detection of Listeria monocytogenes by cross-priming amplification of lmo0733 gene. FEMS Microbiol. Lett., 2014. 361(1): p. 43-51. [CrossRef]
- Long, N., et al., Recent advances and application in whole-genome multiple displacement amplification. Quant. Biol., 2020. 8(4): p. 279-294. [CrossRef]
- Dean, F.B., et al., Comprehensive human genome amplification using multiple displacement amplification. PNAS, 2002. 99(8): p. 5261-5266. [CrossRef]
- Hughes, S., et al., The use of whole genome amplification in the study of human disease. Prog. Biophys. Mol. Biol., 2005. 88(1): p. 173-189. [CrossRef]
- Konakandla, B., Y. Park, and D. Margolies, Whole genome amplification of Chelex-extracted DNA from a single mite: a method for studying genetics of the predatory mite Phytoseiulus persimilis. Exp. Appl. Acarol., 2006. 40(3): p. 241-247. [CrossRef]
- Lepere, C., et al., Whole-genome amplification (WGA) of marine photosynthetic eukaryote populations. FEMS Microbiol. Ecol., 2011. 76(3): p. 513-523. [CrossRef]
- Sabina, J. and J.H. Leamon, Bias in whole genome amplification: causes and considerations. WGA, 2015: p. 15-41. [CrossRef]
- Berthet, N., et al., Phi29 polymerase based random amplification of viral RNA as an alternative to random RT-PCR. BMC Mol. Biol., 2008. 9(1): p. 1-7. [CrossRef]
- Huang, L., et al., Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu. Rev. Genom. Hum. Genet., 2015. 16(1): p. 79-102. [CrossRef]
- Ren, W., C. Wang, and Y. Cai, Loop-mediated isothermal amplification for rapid detection of acute viral necrobiotic virus in scallop Chlamys farreri. Acta Virol., 2009. 53(3): p. 161-167. [CrossRef]
- Jiang, J., et al., Detection of abalone shrivelling syndrome-associated virus using loop-mediated isothermal amplification. J Fish Dis, 2013. [CrossRef]
- Ren, W., et al., Development of a loop-mediated isothermal amplification assay for rapid and sensitive detection of ostreid herpesvirus 1 DNA. J. Virol. Methods, 2010. 170(1-2): p. 30-36. [CrossRef]
- Zaczek Moczydłowska, M.A., et al., A single-tube HNB-based loop-mediated isothermal amplification for the robust detection of the Ostreid herpesvirus 1. Int. J. Mol. Sci., 2020. 21(18): p. 6605. [CrossRef]
- Gao, F., et al., Real-time quantitative isothermal detection of Ostreid herpesvirus-1 DNA in Scapharca subcrenata using recombinase polymerase amplification. J. Virol. Methods, 2018. 255: p. 71-75. [CrossRef]
- Toldrà, A., et al., Detection of isothermally amplified ostreid herpesvirus 1 DNA in Pacific oyster (Crassostrea gigas) using a miniaturised electrochemical biosensor. Talanta, 2020. 207: p. 120308. [CrossRef]
- Xia, J., et al., Complete genome sequence of Ostreid herpesvirus-1 associated with mortalities of Scapharca broughtonii broodstocks. Virol J, 2015. 12: p. 110. [CrossRef]
- Cano, I., et al., Loop-Mediated Isothermal Amplification for the Fast Detection of Bonamia ostreae and Bonamia exitiosa in Flat Oysters. Pathogens, 2024. 13(2): p. 132. [CrossRef]
- Xie, L., et al., Development of a loop-mediated isothermal amplification assay for visual detection of Marteilia refringens in shellfish. Chin. J. Vet. Sci., 2012. 32(7): p. 993-996.
- Mendoza Avilés, I., et al., Loop-mediated isothermal amplification for diagnosing marine pathogens in tissues of Crassostrea spp. and white shrimp, Litopenaeus vannamei, farmed in Mexico. Cienc. Mar., 2021. 47(4): p. 227–239-227–239. [CrossRef]
- Wu, L., et al., Utilization of recombinase polymerase amplification combined with a lateral flow strip for detection of Perkinsus beihaiensis in the oyster Crassostrea hongkongensis. Parasit. Vectors., 2019. 12(1): p. 1-8. [CrossRef]
- Qu, P., et al., Establishment and application of a loop-mediated isothermal amplification (LAMP) method for Perkinsus olseni detection. J. Fish. China. , 2012. 36(8): p. 1281-1289.
- Bass, D., et al., Diverse Applications of Environmental DNA Methods in Parasitology. Trends Parasitol., 2015. 31(10): p. 499-513. [CrossRef]
- Friedman, C.S., et al., Validation of a quantitative PCR assay for detection and quantification of ‘Candidatus Xenohaliotis californiensis’. Dis. Aquat. Organ., 2014. 108(3): p. 251-259. [CrossRef]
- Vincent-Hubert, F., et al., Adsorption of norovirus and ostreid herpesvirus type 1 to polymer membranes for the development of passive samplers. J. Appl. Microbiol., 2017. 122(4): p. 1039-1047. [CrossRef]
- Toldrà, A., et al., Rapid capture and detection of ostreid herpesvirus-1 from Pacific oyster Crassostrea gigas and seawater using magnetic beads. PLoS One, 2018. 13(10): p. e0205207. [CrossRef]
- Morga, B., et al., Haemocytes from Crassostrea gigas and OsHV-1: A promising in vitro system to study host/virus interactions. J. Invertebr. Pathol., 2017. 150: p. 45-53. [CrossRef]
- Sakudo, A. and T. Onodera, Virus capture using anionic polymer-coated magnetic beads. "Int. J. Mol. Med., 2012. 30(1): p. 3-7. [CrossRef]
- Sakudo, A., et al., Capture of dengue virus type 3 using anionic polymer-coated magnetic beads. Int. J. Mol. Med., 2011. 28(4): p. 625-628. [CrossRef]
- Chomczynski, P. and M. Rymaszewski, Alkaline polyethylene glycol-based method for direct PCR from bacteria, eukaryotic tissue samples, and whole blood. Biotechniques, 2006. 40(4): p. 454-458. [CrossRef]
- Dimitrakopoulou, M.-E., et al., Boiling extraction method vs commercial kits for bacterial DNA isolation from food samples. J. Food Nutr. Res., 2020. 3(4): p. 311-319.
- Barreda García, S., et al., Helicase-dependent isothermal amplification: a novel tool in the development of molecular-based analytical systems for rapid pathogen detection. Anal. Bioanal. Chem., 2018. 410(3): p. 679-693. [CrossRef]
- Nagamine, K., T. Hase, and T. Notomi, Accelerated reaction by loop-mediated isothermal amplification using loop primers. MCP, 2002. 16(3): p. 223-229. [CrossRef]
- Yi, T.t., et al., Betaine-assisted recombinase polymerase assay for rapid hepatitis B virus detection. Biotechnol. Appl. Biochem., 2021. 68(3): p. 469-475. [CrossRef]
- Pasookhush, P., et al., Development of duplex loop-mediated isothermal amplification (dLAMP) combined with lateral flow dipstick (LFD) for the rapid and specific detection of Vibrio vulnificus and V. parahaemolyticus. N. Am. J. Aquac., 2016. 78(4): p. 327-336. [CrossRef]
- Surasilp, T., et al., Rapid and sensitive detection of Vibrio vulnificus by loop-mediated isothermal amplification combined with lateral flow dipstick targeted to rpoS gene. MCP, 2011. 25(4): p. 158-163. [CrossRef]
- Chu, F.L. and R. Hale, Relationship between pollution and susceptibility to infectious disease in the eastern oyster, Crassostrea virginica. Mar. Environ. Res., 1994. 38(4): p. 243-256. [CrossRef]
- Renault, T., Viruses infecting marine mollusks Studies in viral ecol., 2021: p. 275-303.
Figure 1.
The world molluscs production by type and geographical location. A) The production volume of major mollusc species in 2018, 2020 and 2022. B) Molluscs-producing countries worldwide and the break-down of their production ratios [4].
Figure 1.
The world molluscs production by type and geographical location. A) The production volume of major mollusc species in 2018, 2020 and 2022. B) Molluscs-producing countries worldwide and the break-down of their production ratios [4].
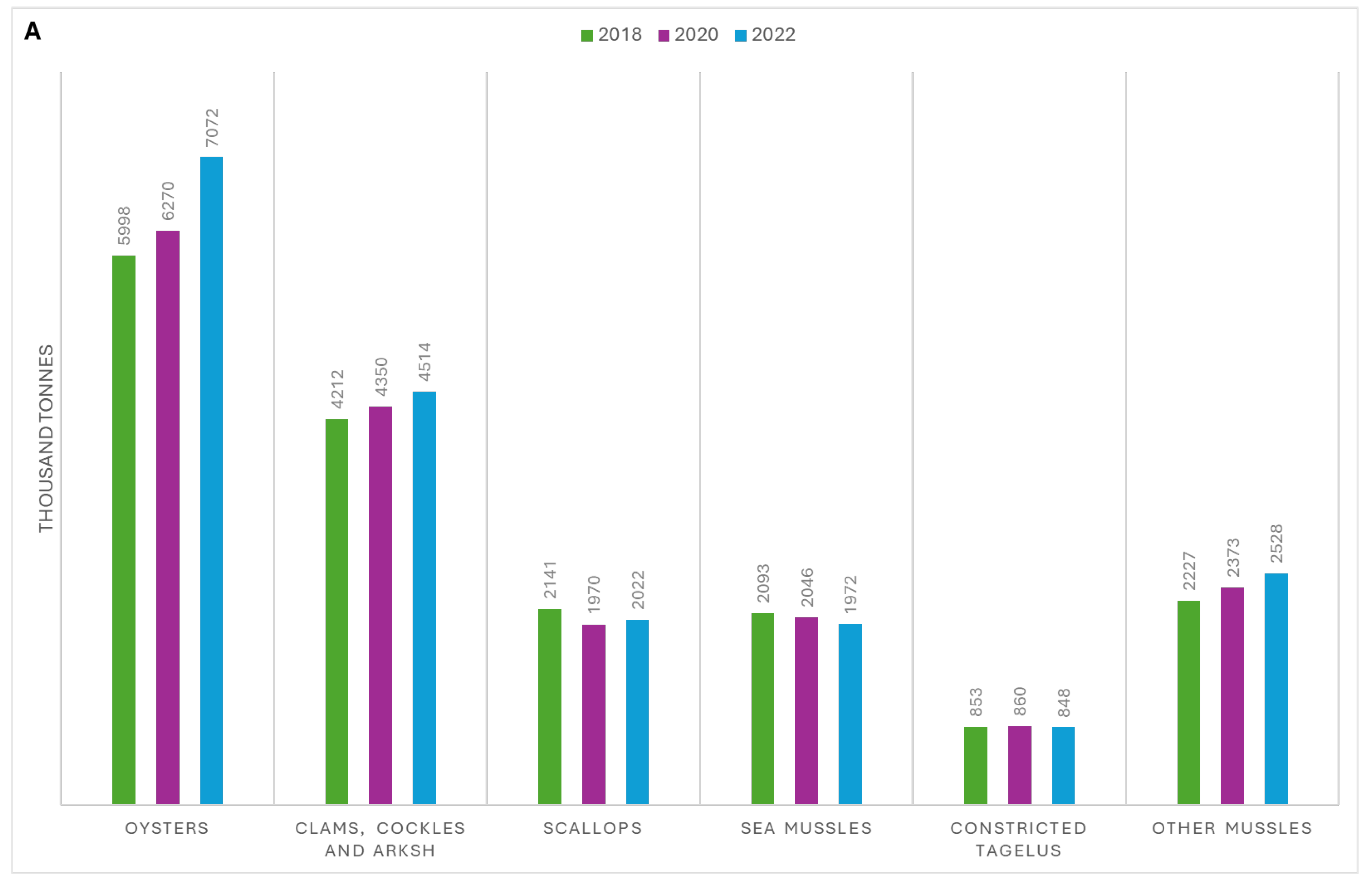
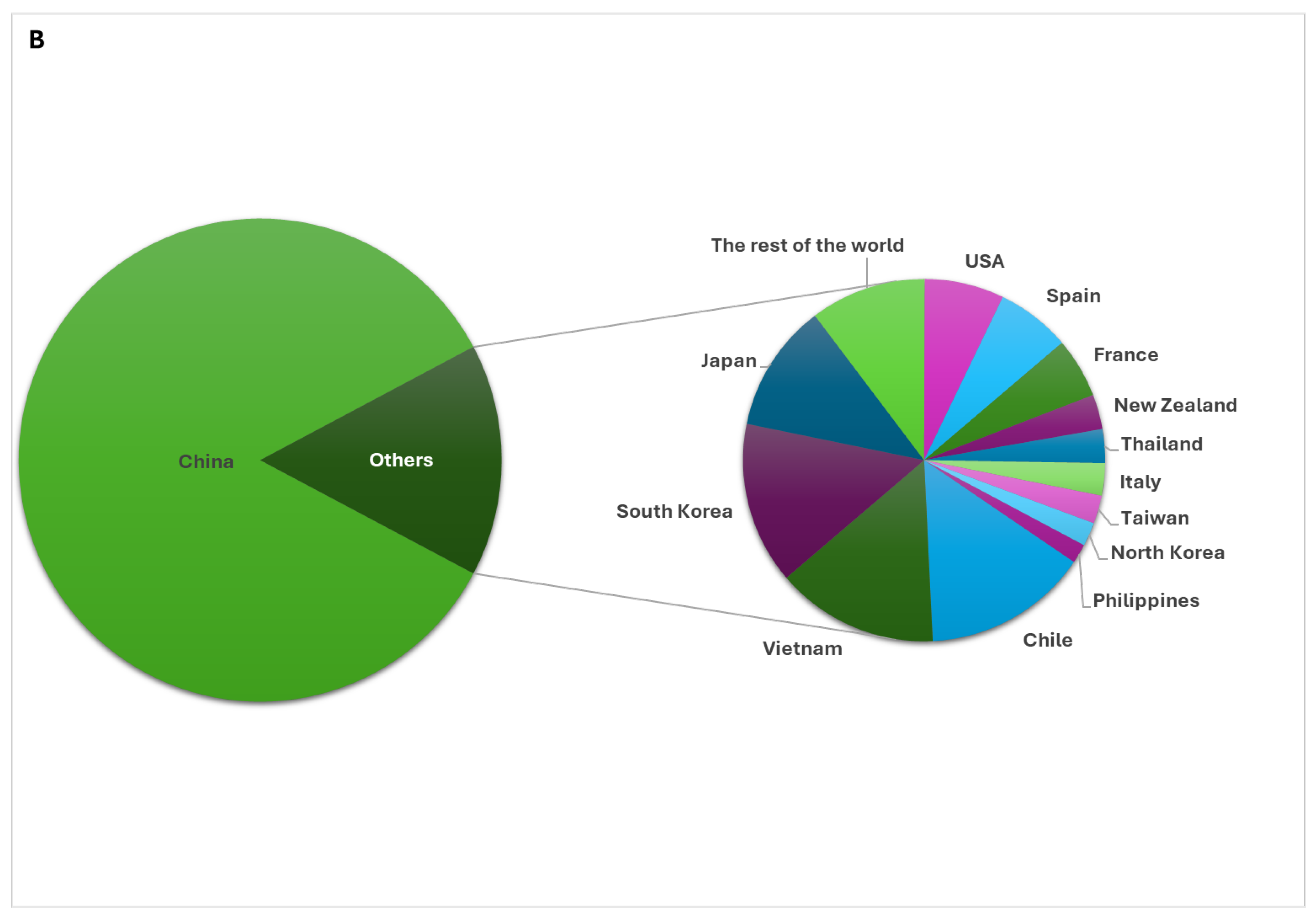
Figure 2.
Comparison between the amplification mechanism for loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), cross-priming amplification (CPA) and multiple displacement amplification (MDA). A) LAMP, A1) The annealing of inner primers (FIP/BIP) to the target strand and elongation with the isothermal polymerase (orange blob), then displacement of the new strands by the outer primers (F3/B3). A2) The newly amplified strands with joined F1c and B1c annealed to their complimentary regions on the same strand forming dumbbell shaped DNA. A3) Multiple amplifications and strand displacements by inner and loop primers resulting in loop and stem-like structures. B) RPA, B1) Recombinase protein (red strips)-primer complex with each of the forward (pink) or reverse (green) primer hybridize to the target sequence on sense and antisense strand, respectively, with the now free single stranded DNA stabilized by single stranded binding proteins (yellow circles). B2) Following complex disassembly, elongation of the new strand is initiated by polymerase DNA (purple blob). B3) The reaction continues with the new double strands serve as templates for the recombinase-primer complex. C) CPA C1) Half of the cross primer anneals to the complementary region on the target strand then extension occurs using the polymerase (light red). The generated strand with flanking sequence to the 5’ end allows it to be displaced by the outer forward primer (dark grey). C2) A long and shorter products are generated. The longer amplification products has the cross priming complementary region (pink) to another sequence on the same strand C3) The formation of the hair pin structure from the annealing of the complementary region of the cross primer and the complementary sequence on the same strand while the short formed strand cannot produce the hairpin structure. C4) Newly formed hair pin structures serves as template for primers and produce more amplicons. D) Whole genomic amplification (WGA)-MDA D1) Many random hexamer primers (pink) hybridize to the template and extend using phi29 DNA polymerase (blue blob). D2) Occurrence of strand displacement by the new extending primers. D3) Continuous amplification of the newly created strands result in the formation of networks of branched DNA structures. Created in BioRender. Abbas, H. (2025) https://BioRender.com/w75z807.
Figure 2.
Comparison between the amplification mechanism for loop-mediated isothermal amplification (LAMP), recombinase polymerase amplification (RPA), cross-priming amplification (CPA) and multiple displacement amplification (MDA). A) LAMP, A1) The annealing of inner primers (FIP/BIP) to the target strand and elongation with the isothermal polymerase (orange blob), then displacement of the new strands by the outer primers (F3/B3). A2) The newly amplified strands with joined F1c and B1c annealed to their complimentary regions on the same strand forming dumbbell shaped DNA. A3) Multiple amplifications and strand displacements by inner and loop primers resulting in loop and stem-like structures. B) RPA, B1) Recombinase protein (red strips)-primer complex with each of the forward (pink) or reverse (green) primer hybridize to the target sequence on sense and antisense strand, respectively, with the now free single stranded DNA stabilized by single stranded binding proteins (yellow circles). B2) Following complex disassembly, elongation of the new strand is initiated by polymerase DNA (purple blob). B3) The reaction continues with the new double strands serve as templates for the recombinase-primer complex. C) CPA C1) Half of the cross primer anneals to the complementary region on the target strand then extension occurs using the polymerase (light red). The generated strand with flanking sequence to the 5’ end allows it to be displaced by the outer forward primer (dark grey). C2) A long and shorter products are generated. The longer amplification products has the cross priming complementary region (pink) to another sequence on the same strand C3) The formation of the hair pin structure from the annealing of the complementary region of the cross primer and the complementary sequence on the same strand while the short formed strand cannot produce the hairpin structure. C4) Newly formed hair pin structures serves as template for primers and produce more amplicons. D) Whole genomic amplification (WGA)-MDA D1) Many random hexamer primers (pink) hybridize to the template and extend using phi29 DNA polymerase (blue blob). D2) Occurrence of strand displacement by the new extending primers. D3) Continuous amplification of the newly created strands result in the formation of networks of branched DNA structures. Created in BioRender. Abbas, H. (2025) https://BioRender.com/w75z807.
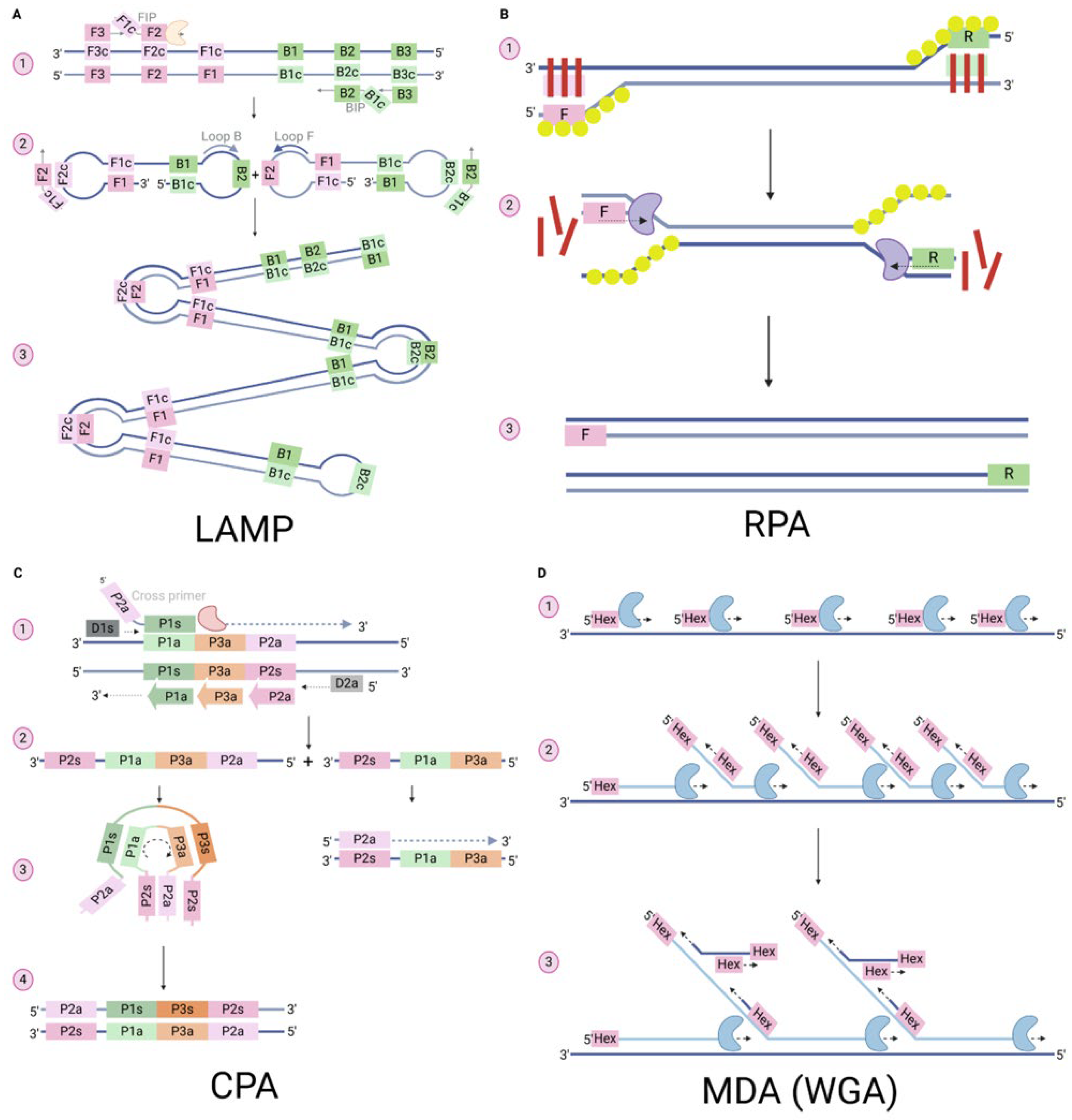
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



