
Submitted:
13 April 2025
Posted:
14 April 2025
You are already at the latest version
Abstract
Prion diseases are typically attributed to the accumulation of protease-resistant prion protein (PrPSc), yet growing evidence indicates that alternative misfolded PrP conformers and inflammatory factors may also drive neurodegeneration. Whether bac-terial lipopolysaccharide (LPS) can independently generate or exacerbate prion-like pathology in the absence of classical PrPSc remains unclear. This study aimed to determine if recombinant moPrPRes, produced by incubating normal mouse PrPC with LPS, induces prion-like disease in wild-type FVB/N mice, whether LPS alone triggers neurodegen-eration, and how co-administration of LPS influences moPrPRes and RML prion pathology. FVB/N mice were randomized into six subcutaneous treatment groups: saline, LPS, moPrPRes, moPrPRes+LPS, RML, or RML+LPS. Animals were monitored for survival, clinical signs, and body weight. Brains were assessed histologically and biochemically for spongiform changes, astrogliosis, and protease-resistant PrP. Subcutaneous moPrPRes induced a distinctive encephalopathy, with 20% mortality by 200 days post-inoculation (dpi), rising to 60% by study’s end, despite undetectable PrPSc. Chronic LPS infusion alone caused an Alzheimer’s-like pathology (~40% mortality). Co-administration of moPrPRes with LPS resulted in 30% mortality by 200 dpi and 50% survival at 750 dpi. Keywords: keyword 1; keyword 2; keyword 3 (List three to ten pertinent keywords specific to the article yet reasonably common within the subject discipline.) Notably, LPS strongly synergized with RML, accelerating disease onset, intensifying PrP deposition, and ex-acerbating spongiform changes compared to RML alone. Instead, misfolded prion con-formers and endotoxin-driven inflammation can independently or synergistically pro-mote neuropathology. Targeting inflammatory pathways and subclinical prion con-formers may thus provide new therapeutic avenues for prion-like disorders.
Keywords:
recombinant prion protein
; lipopolysaccharide
; cofactor
; neurodegeneration
; prion disease
1. Introduction
Prion diseases, also referred to as transmissible spongiform encephalopathies (TSE), occupy a unique position among neurodegenerative conditions due to their infectious nature and the central role played by misfolded scrapie prion protein (PrPSc) [1,2]. According to the “protein-only” hypothesis, prion pathogenesis arises when the normally soluble cellular prion protein (PrPC) adopts a pathogenic β-sheet-rich conformation (PrPSc) capable of templating the misfolding of additional PrPC molecules [1]. This self-propagating mechanism has profoundly influenced how researchers conceive of other protein misfolding disorders, including Alzheimer’s disease, Parkinson’s disease, and bovine spongiform encephalopathy, all of which appear to share pathological features such as aberrant protein conformers and progressive neuronal degeneration [3,4]. Despite these conceptual advances, significant gaps persist in our understanding of precisely how prion-like mechanisms intersect with inflammatory processes, particularly those driven by bacterial components, during the onset and progression of brain neurodegeneration.
Emerging evidence suggests that bacterial lipopolysaccharide (LPS), a key molecule from the outer membrane of Gram-negative bacteria, might contribute to or exacerbate prion-like processes. In vitro studies have demonstrated that LPS can induce the formation of protease-resistant recombinant prion protein (moPrPRes) [5,6], raising intriguing possibilities about the ability of bacterial factors to modulate pathological protein folding in vivo. Moreover, chronic peripheral administration of LPS has been shown to exacerbate the pathophysiology of other neurodegenerative proteinopathies, including Alzheimer’s disease and Parkinson’s disease [7,8,9,10,11]. In these conditions, LPS activates the immune system [12,13], leading to microglial overactivation and sustained neuroinflammation within the central nervous system (CNS), key processes that further promote neuronal damage [14,15,16]. Indeed, prolonged peripheral exposure to LPS can compromise the blood–brain barrier, alter cytokine networks, and potentiate protein misfolding, thereby driving or exacerbating neurodegenerative pathology [15,16,17,18,19].
Traditionally, experimental models of scrapie rely on intracerebral inoculation because it yields relatively short incubation periods. However, to better mimic natural prion transmission, alternative peripheral routes, such as subcutaneous injection, have also been used, even though they typically require longer incubation times [12,21]. Importantly, subcutaneous administration has been shown to be equally effective for disease transmission in various animal models and may provide a more physiologically relevant approximation of how prions disseminate from peripheral tissues to the CNS [12,21].
Whether LPS alone, or in combination with a prion-like substrate, can trigger or accelerate prion-like neurodegeneration in healthy animals remains unclear. Addressing this gap is crucial because it could resolve whether misfolded prion protein isoforms indeed require additional factors such as LPS to exert their full pathogenic effects in vivo, or whether prion-like neurodegeneration can be wholly attributed to misfolded protein conformers. A clearer delineation of these processes is particularly important given the rapidly expanding recognition that many neurodegenerative syndromes likely exist on a spectrum of protein misfolding and inflammatory pathologies, rather than being wholly distinct entities.
Thus, in this study, we aimed to determine whether LPS-converted recombinant prion protein (moPrPRes), generated entirely in vitro by incubation with Escherichia coli 0111:B4 LPS, without traditional amplification methods such as seeding or serial protein misfolding cyclic amplification (sPMCA), could independently induce prion-like neurodegeneration in wild-type mice following subcutaneous administration. Additionally, we sought to assess whether chronic peripheral exposure to bacterial LPS, in the absence of classical prion agents, might trigger spongiform brain pathology and associated neurodegenerative changes. Finally, we investigated the synergistic effects of co-administering LPS with a classical scrapie strain (Rocky Mountain Laboratory, RML) and moPrPRes, focusing on its impact on disease progression, neuropathological severity, and prion protein deposition. By exploring these mechanisms, our study aims to investigate the complex interactions between inflammatory triggers and prion-like processes, shedding light on how these interactions may contribute to the early stages of prion and other protein-misfolding neurodegenerative disorders.
This section is not mandatory but may be added if there are patents resulting from the work reported in this manuscript.
2. Results
2.1. Weight Change
Throughout the experiment, animals from all treatment groups were weighed monthly, starting at 6 weeks of age until termination at 74, 102, and 110 weeks. This monitoring allowed for the observation of weight trends over time across the different treatment groups (Figure 1). Statistical comparisons revealed significant differences (p < 0.05) between most treatment groups at various time points. Notably, significant differences were observed between the LPS and RML treatments at 35, 39, 43, 48, 52, and 66 weeks; between LPS and saline treatments at 48, 52, 56, 66, and 110 weeks; between LPS and RML+LPS treatments at 35 and 39 weeks; between LPS and moPrPres treatments at 43, 48, 52, 56, 61, 66, 70, and 74 weeks; and between LPS and moPrPres+LPS treatments at 39, 43, 48, 52, 56, 61, 66, 70, 74, 102, and 110 weeks.
The average weight trend indicates that LPS-treated animals consistently exhibited higher body weights than all other treatment groups starting at 30 weeks of age. Statistically significant weight differences (p < 0.05) between the LPS treatment group and the saline, moPrPres, moPrPres+LPS, and RML groups were observed at 48, 52, and 66 weeks. Furthermore, the LPS-treated group showed significantly greater weights than the moPrPRes and moPrPRes+LPS groups at 61, 70, and 74 weeks of age.
The LPS+RML treatment group experienced a marked decline in weight from 30 to 39 weeks of age. This decline was associated with early mortality, accompanied by typical clinical signs of prion disease in all animal replicates within this treatment group. The moPrPRes-treated group exhibited significantly higher weights than the RML and RML+LPS groups at 35 and 39 weeks of age, respectively. However, the mean weight of the moPrPres group remained significantly lower than that of the LPS-treated group at 43, 48, 52, 56, 61, 66, 70, and 74 weeks of age. Both the moPrPres and RML-treated groups consistently displayed lower average body weights compared to the saline negative control group, starting from 30 weeks onward.
2.2. Survival Analysis
At 11 weeks post-infection (wpi), five mice from each treatment group were randomly euthanized (Figure 2). At this point, none of the mice exhibited clinical signs of prion disease or any abnormalities. The remaining ten mice in each treatment group were monitored until they developed typical clinical manifestations of prion disease, which included kyphosis, ataxia, dysmetria, tremors, head tilt, tail rigidity, bradykinesia, proprioceptive deficits, stupor, loss of deep pain sensation, and significant weight loss over a period of more than 72 hours.
In the RML treatment group, there was a substantial decline in the number of mice, with an 80% drop observed after 200 days post-infection (dpi). However, two mice survived until close to the end of the experiment at 700 dpi, resulting in a survival rate of 10%. In contrast, the negative saline control group maintained a 100% survival rate for over 600 dpi but exhibited a 40% mortality rate just one month prior to the experiment's termination at 750 dpi.
The RML+LPS treatment group experienced a 30% loss of experimental animals after 100 dpi, with a sudden increase to 100% mortality by 200 dpi. Conversely, during the first 350 dpi, the LPS-treated mice had a lower mortality rate of 10%. However, deaths in this group increased to 40% after 650 dpi, resulting in a survival rate of 60% at the termination of the experiment.
The moPrPRes treatment group showed a 20% loss of animals by 200 dpi, which escalated to a 60% mortality rate close to the experiment's conclusion. Nevertheless, 40% of the moPrPRes-treated animals survived until 750 dpi. The moPrPRes+LPS treatment group experienced a 30% reduction in its population within the first 200 dpi and concluded with a survival rate of 50% at the end of the study.
2.3. Immunohistochemical Staining
2.3.1. Lipopolysaccharide-Only Treated Animals
Five mice treated exclusively with LPS and euthanized at 11 weeks post-infection (wpi), with no apparent clinical signs of disease, exhibited mild vacuolation in the cerebral cortex (Cc), thalamus (Th), midbrain (Mb), and cerebellum (Cr) (Figure 3.B, e-h). The corresponding PrPSc staining in these brain regions showed minimal differences compared to the saline-treated control group (Figure 5, a-d).
In contrast, LPS-only treated FVB/N mice that reached the terminal stage of illness demonstrated widespread vacuolation in the Cc, Th, Mb, and particularly the Cr (Figure 4, e-h). The vacuolation pattern observed in these terminally sick mice was comparable to that seen in the positive control group (RML) at the terminal stage, indicating similar vacuole distribution across various brain regions. However, the total number of vacuoles was lower in the terminally sick LPS-only treated animals compared to the RML group.
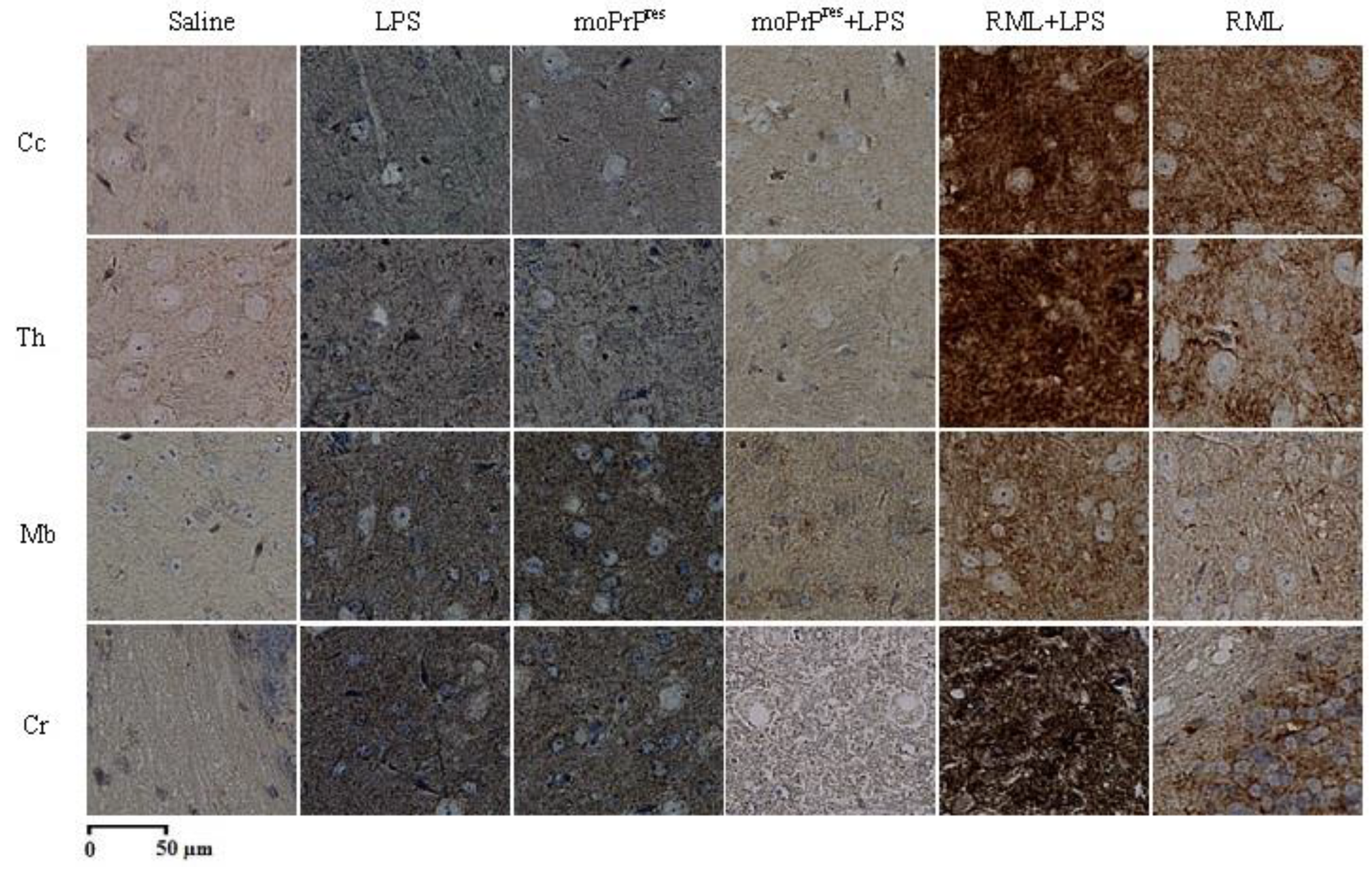
Results of the PrPSc staining in terminally sick LPS-only treated mice (Figure 6, e-h) revealed significant differences in PrPSc deposition across brain regions when compared to the positive controls (Figure 6, u-x). Notably, the LPS-treated animals exhibited no deposits of the pathogenic prion protein (PrPSc) in any brain regions (Figure 6, e-h).
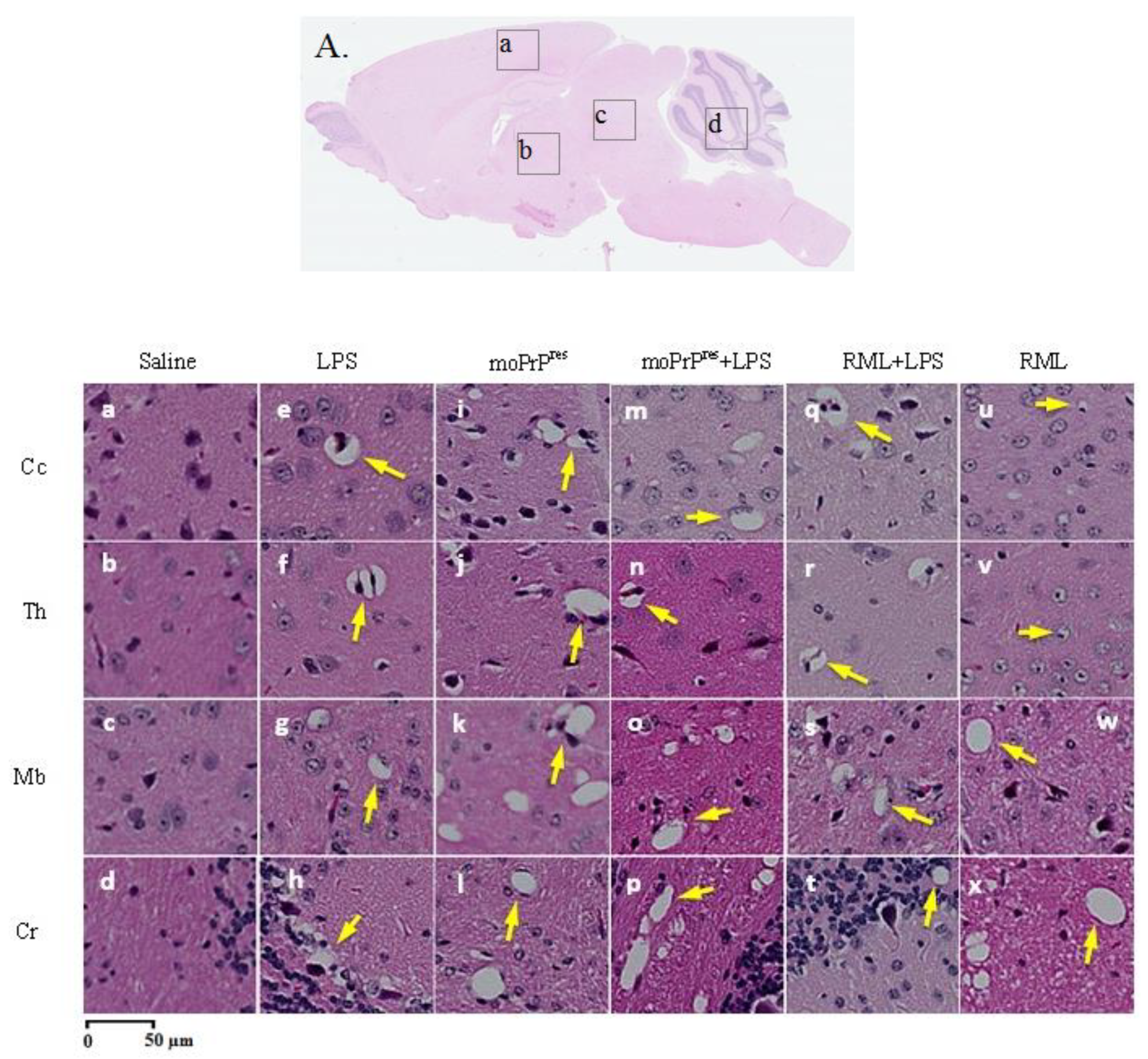
Figure 3.
Early demonstration of minor prion-like vacuolation in hematoxylin and eosin staining of the brains of FVB/N wild-type female mice (10X magnification). Mice treated with subcutaneous administration of bacterial LPS from Escherichia coli 0111 for 6 weeks (B, e-h), moPrPres (B, i-l), moPrPres+LPS (B, m-p), and RML+LPS (B, q-t), and euthanized at 11 weeks post-inoculation exhibited minor vacuolation compared to positive (B, u-x) and negative (B, a-d) controls (RML and saline treatment groups, respectively) in various brain regions (A) including the cerebral cortex (Cc; A.a), thalamus (Th; A.b), midbrain (Mb; A.c), and cerebellum (Cr; A.d).
Figure 3.
Early demonstration of minor prion-like vacuolation in hematoxylin and eosin staining of the brains of FVB/N wild-type female mice (10X magnification). Mice treated with subcutaneous administration of bacterial LPS from Escherichia coli 0111 for 6 weeks (B, e-h), moPrPres (B, i-l), moPrPres+LPS (B, m-p), and RML+LPS (B, q-t), and euthanized at 11 weeks post-inoculation exhibited minor vacuolation compared to positive (B, u-x) and negative (B, a-d) controls (RML and saline treatment groups, respectively) in various brain regions (A) including the cerebral cortex (Cc; A.a), thalamus (Th; A.b), midbrain (Mb; A.c), and cerebellum (Cr; A.d).
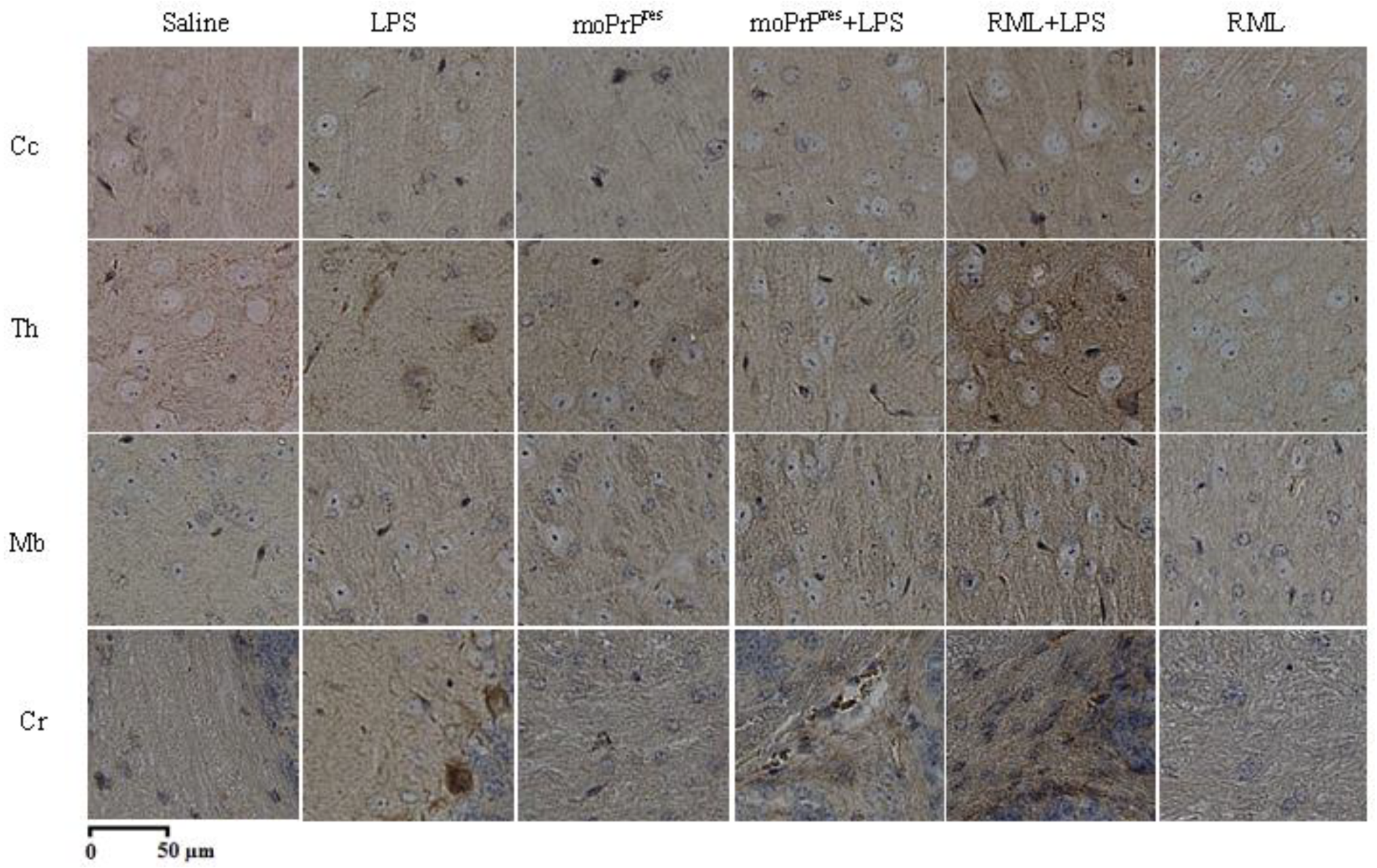
Additionally, terminally sick LPS-only treated mice displayed aggravated astrogliosis specifically in the Cr region, akin to the RML group (Figure 7, h and x). In contrast, other brain regions, including the Cc, Th, and Mb, showed no significant differences compared to the saline group (Figure 7, a-g).
This pattern was also observed in amyloid plaque (Ap) staining, where only the Cr region of terminally sick LPS-treated mice exhibited amyloid plaques (Figure 8, h). This level of Ap accumulation in the Cr was distinctive, as none of the other treatment groups or controls demonstrated similar Ap deposition in this region (Figure 8, d, l, p, and x).
2.3.2. Lipopolysaccharide-Converted moPrPres Treated Groups
Mice treated with recombinant moPrPres and euthanized at 11 weeks post-infection (wpi), with no observable clinical abnormalities, exhibited mild vacuolation in several brain regions, including the Cc, Th, Mb, and Cr (Figure 3.B, i-l). In comparison, the negative controls (Figure 3.B, a-d) displayed either a complete absence of vacuolation or only slight vacuolation in similar brain regions. The RML-treated groups (Figure 3.B, u-x) exhibited similar findings at 11 wpi, showing slight vacuolation.
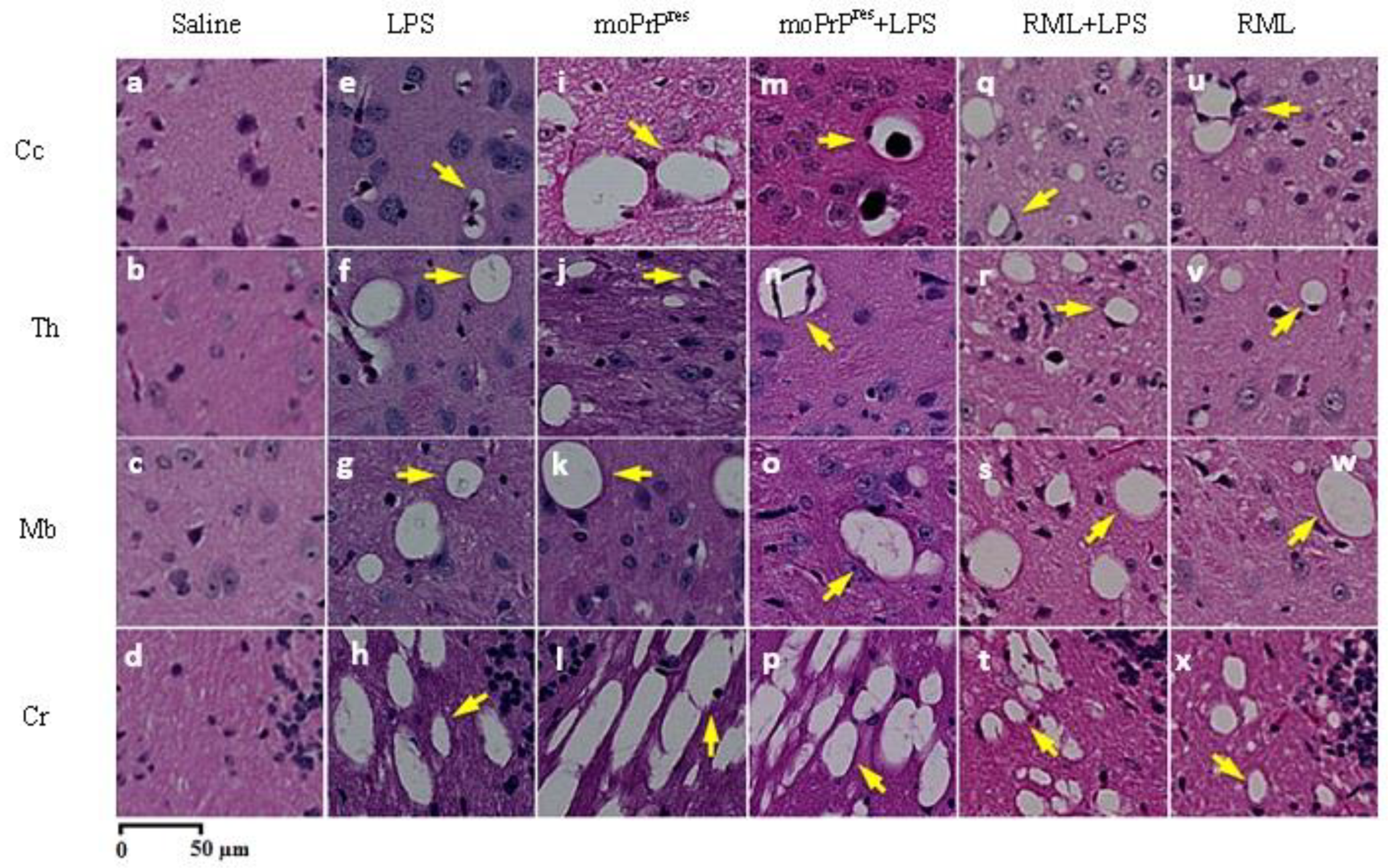
Figure 4.
Prion-like vacuolation profile in hematoxylin and eosin staining of brains from terminally sick FVB/N wild-type female mice (10X magnification). Mice euthanized or found dead with terminal prion-like clinical signs and treated with subcutaneous LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), moPrPRes+LPS (m-p), and RML+LPS (q-t) demonstrated major vacuolation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions, including Cc, Th, Mb, and Cr. The moPrPRes-containing and LPS treatment groups exhibited larger vacuoles compared to the RML-containing groups.
Figure 4.
Prion-like vacuolation profile in hematoxylin and eosin staining of brains from terminally sick FVB/N wild-type female mice (10X magnification). Mice euthanized or found dead with terminal prion-like clinical signs and treated with subcutaneous LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), moPrPRes+LPS (m-p), and RML+LPS (q-t) demonstrated major vacuolation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions, including Cc, Th, Mb, and Cr. The moPrPRes-containing and LPS treatment groups exhibited larger vacuoles compared to the RML-containing groups.
Furthermore, PrPSc staining in the brains of moPrPres-treated mice at 11 weeks post-infection (Figure 5, i-l) revealed no detectable PrPSc deposits, contrasting with the saline-treated negative controls (Figure 5, a-d) and the RML-treated group (Figure 5, u-x), which displayed significant PrPSc accumulation at the same time point.
Results from hematoxylin and eosin (H&E) staining of terminally sick moPrPRes-treated mice indicated notable vacuolation across various brain regions, particularly pronounced in the Cr (Figure 4, i-l). The distribution of vacuolation in the brains of these terminally sick mice bore a clear resemblance to that observed in terminally sick RML control mice; however, the moPrPRes-treated group exhibited lower vacuole abundance in the Cc and larger vacuole sizes in the Cc, Th, Mb, and Cr regions (Figure 4, i-l).
Additionally, PrPSc staining results for terminally sick moPrPRes-treated animals showed no accumulation of PrPSc in the Cc, Th, Mb, or Cr (Figure 6, i-l).
Moreover, moPrPRes-treated animals displayed slight astrogliosis in the Cr (Figure 7, l) when compared to the RML positive controls. Mild astrogliosis was also observed in the Mb of this treatment group (Figure 7, k), differentiating it from the negative controls. However, the Cc and Th regions of the moPrPRes treatment group did not exhibit astrogliosis compared to the corresponding regions in the negative controls (Figure 7, i-j). Importantly, no Ap deposition was detected in any of the brain regions examined in this group of mice (Figure 8, i-l).
Figure 5.
PrPSc deposition in brains of FVB/N wild-type female mice at 11 weeks post-inoculation (10X magnification). Mice euthanized at 11 weeks post-inoculation treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes + LPS (m-p) exhibited no PrPSc deposition, whereas the RML + LPS treatment (q-t) showed PrPSc accumulation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions including Cc, Th, Mb, and Cr.
Figure 5.
PrPSc deposition in brains of FVB/N wild-type female mice at 11 weeks post-inoculation (10X magnification). Mice euthanized at 11 weeks post-inoculation treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes + LPS (m-p) exhibited no PrPSc deposition, whereas the RML + LPS treatment (q-t) showed PrPSc accumulation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions including Cc, Th, Mb, and Cr.
2.3.3. Lipopolysaccharide-Converted moPrPres and LPS Treatment Group
At 11 weeks post-infection (wpi), five FVB/N female mice from the moPrPRes+LPS treatment group, which exhibited no clinical signs of disease, were randomly selected for euthanasia. Histological examination using hematoxylin and eosin (H&E) staining of their brain slides revealed mild vacuolation across multiple brain regions, including the Cc, Th, Mb, and Cr (Figure 3.B, m-p). Notably, there was an absence of PrPSc staining in this group at this time point (Figure 3, m-p).
In terminally sick mice from the moPrPRes+LPS treatment group, widespread vacuolation was observed in the Cc, Th, Mb, and Cr regions (Figure 4, m-p), which was comparable to findings in the terminally sick RML group (Figure 4, u-x). However, the number of vacuoles in the moPrPRes+LPS-treated animals was lower in equivalent brain regions compared to the terminally sick RML-treated mice. Similar to the observations in the LPS and moPrPRes treatment groups, terminally sick moPrPRes+LPS-treated mice exhibited larger vacuoles when compared to those in the RML-treated group. Importantly, terminally sick moPrPRes+LPS-treated mice showed no accumulation of PrPSc (Figure 6, m-p), consistent with the negative saline-treated controls (Figure 6, a-d).
Immunohistochemical (IHC) staining in this treatment group revealed mild astrogliosis across all brain regions, with the Cr region displaying the most pronounced effects (Figure 7, m-p). In comparison to the negative controls, moPrPres+LPS-treated animals exhibited significant differences in the Cc, Th, and Mb, although only a few instances of astrogliosis were noted in these areas (Figure 7, m-o). Conversely, the positive controls showed severe astrogliosis in similar brain regions (Figure 7, u-w) when compared to the moPrPres+LPS treatment group. While the Cr region exhibited more intense astrogliosis compared to the other three brain regions in the moPrPres+LPS-treated animals, it was still less severe than that observed in the positive controls (Figure 7, p and x).
Additionally, Ap staining in the moPrPRes+LPS treatment group revealed only mild Ap deposition in the Th, with no signs of Ap accumulation detected in the other brain regions (Figure 8, m-p).
Figure 6.
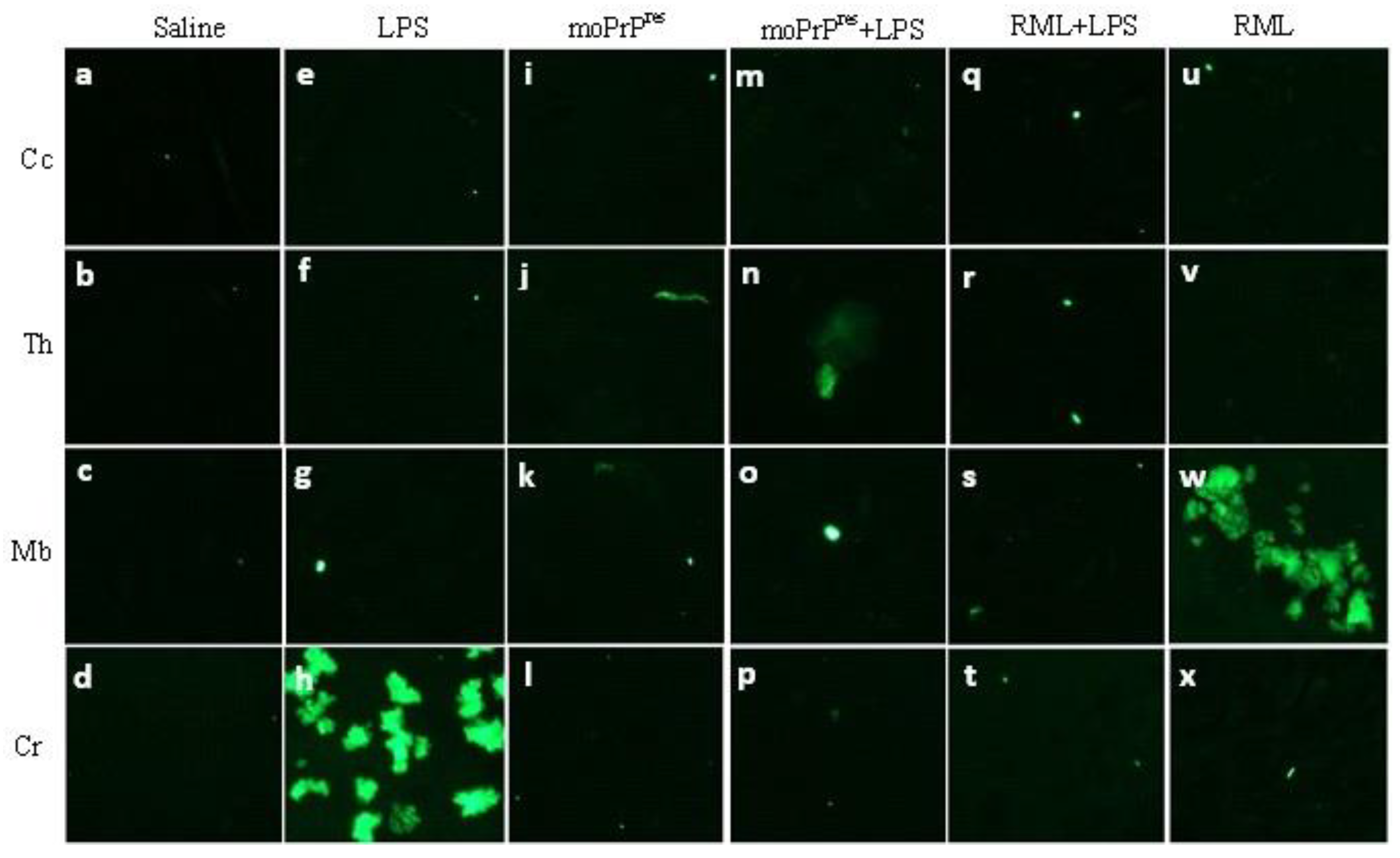
Prion protein scrapie (PrPSc) deposition in brains of FVB/N wild-type female mice at terminal sickness (10X magnification). Mice euthanized at terminal sickness treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) showed no PrPSc deposition (dark brown color staining), whereas the RML+LPS treatment (q-t) exhibited aggravated PrPSc accumulation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions, including Cc, Th, Mb, and Cr.
Figure 6.
Prion protein scrapie (PrPSc) deposition in brains of FVB/N wild-type female mice at terminal sickness (10X magnification). Mice euthanized at terminal sickness treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) showed no PrPSc deposition (dark brown color staining), whereas the RML+LPS treatment (q-t) exhibited aggravated PrPSc accumulation compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) in various brain regions, including Cc, Th, Mb, and Cr.
2.3.4. Rocky Mountain Laboratory+LPS Treatment Group
Mice treated with RML+LPS and euthanized at 11 weeks post-infection (wpi), with no clinical signs of prion disease, exhibited mild vacuolation in various brain regions, including the Cc, Th, Mb, and Cr (Figure 3.B, q-t). Notably, PrPSc accumulation in these brain regions demonstrated subjective differences when compared to RML-treated mice at 11 wpi (Figure 5, u-x), with the RML+LPS group exhibiting greater intensity of PrPSc accumulation (Figure 5, q-t).
In terminally sick mice treated with RML+LPS, widespread vacuolation was observed in the Cc, Th, Mb, and Cr regions (Figure 4, q-t). The distribution of vacuolation in this group was similar to that seen in the terminally sick RML positive controls, with a greater intensity of vacuolation in corresponding brain regions. A comparison between terminally sick RML+LPS-treated animals (Figure 6, q-t) and the RML positive controls (Figure 6, u-x) revealed distinct differences in PrPSc accumulation, particularly in the Cc, Th, and Cr.
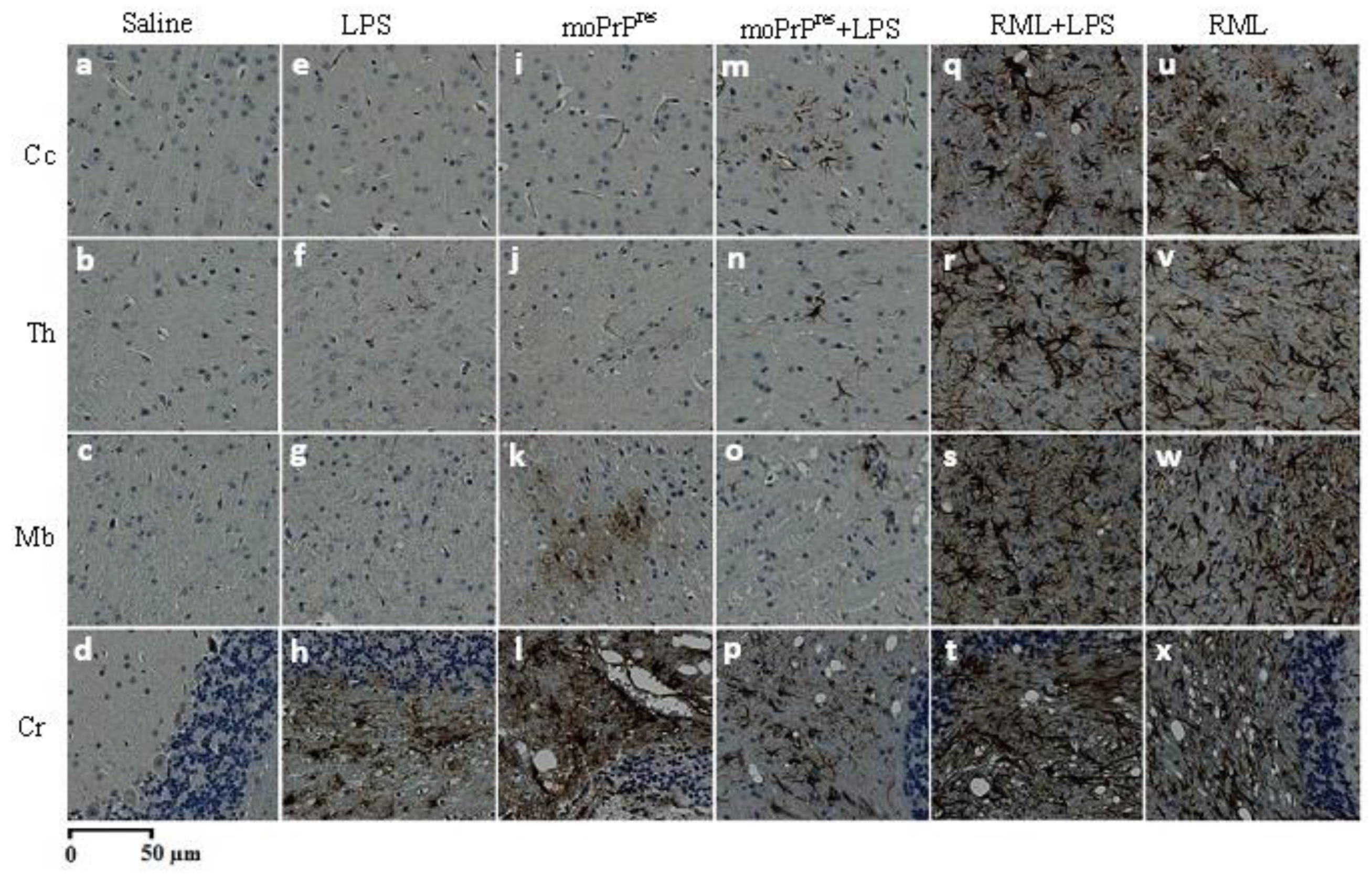
Astrogliosis staining of the RML+LPS treatment group (Figure 7, q-t) demonstrated significant astrogliosis across all brain regions, showing greater severity than that observed in the RML-alone treated mice (Figure 7, u-x). The brains of RML+LPS-treated mice exhibited more pronounced astrogliosis than those of the RML-treated group. However, staining for Ap showed no differences between the RML and RML+LPS-treated mice (Figure 8, q-x); both groups lacked Ap deposition in all brain regions except for the Mb in the RML-treated mice (Figure 8, w).
Figure 7.
Astrogliosis in brains of FVB/N wild-type female mice. Mice euthanized at terminal sickness treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) exhibited minor astrogliosis, with only the cerebellum (Cr) region in moPrPRes-treated groups showing aggravated astrogliosis. Conversely, the RML+LPS treatment (q-t) displayed significant astrogliosis compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) across various brain regions, including Cc, Th, Mb, and Cr.
Figure 7.
Astrogliosis in brains of FVB/N wild-type female mice. Mice euthanized at terminal sickness treated with subcutaneous administration of LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) exhibited minor astrogliosis, with only the cerebellum (Cr) region in moPrPRes-treated groups showing aggravated astrogliosis. Conversely, the RML+LPS treatment (q-t) displayed significant astrogliosis compared to positive (u-x) and negative (a-d) controls (RML and saline treatment groups, respectively) across various brain regions, including Cc, Th, Mb, and Cr.
2.3.5. Comparison of LPS-Treated and Non-LPS-Treated Mice
At 11 wpi, H&E staining showed few differences between the LPS+RML and RML treatment groups, with both groups displaying minimal vacuolation in most brain regions. This pattern was similarly observed in the moPrPRes-treated groups, where only minor subjective differences in vacuolation were noted between the moPrPRes and moPrPRes+LPS treatment groups. PrPSc deposition patterns were also comparable between the LPS-treated and non-LPS-treated groups; however, both moPrPRes-treated groups displayed similar results to the negative controls at 11 wpi. In contrast, both the moPrPRes and RML-treated groups exhibited clear differences from the negative saline control group in the H&E slides at this time point.
In terminally sick mice, the combination of LPS with RML appeared to increase the abundance and distribution of vacuolation in the brain compared to the RML-only treatment group. Widespread vacuolation was evident in the H&E staining of the terminally sick RML+LPS treatment group across all examined brain regions, including the Cc, corpus callosum, hippocampus, Th, hypothalamus, Mb, and Cr. In contrast, terminally sick RML-treated mice exhibited lower vacuolation intensity in similar brain regions.
Figure 8.
Staining for amyloid plaques in brains of FVB/N wild-type female mice. Mice euthanized at terminal sickness treated with subcutaneous LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) showed no amyloid plaque (Ap) deposition, with only the cerebellum (Cr) region in the LPS-treated group demonstrating accumulation. A similar pattern was observed in the RML+LPS treatment (q-t) and the RML treatment group (u-x), with Ap deposition occurring solely in the Cr of RML-treated mice.
Figure 8.
Staining for amyloid plaques in brains of FVB/N wild-type female mice. Mice euthanized at terminal sickness treated with subcutaneous LPS from Escherichia coli 0111:B4 for 6 weeks (e-h), moPrPRes (i-l), and moPrPRes+LPS (m-p) showed no amyloid plaque (Ap) deposition, with only the cerebellum (Cr) region in the LPS-treated group demonstrating accumulation. A similar pattern was observed in the RML+LPS treatment (q-t) and the RML treatment group (u-x), with Ap deposition occurring solely in the Cr of RML-treated mice.
Similar trends were observed in the results of PrPSc and astrogliosis staining. Terminally sick RML+LPS-treated mice displayed a subjectively greater intensity of PrPSc accumulation in the Cc and Th, while terminally sick RML-treated mice showed slightly lower PrPSc accumulation in these regions (Figure 6, q-x). This pattern was also reflected in astrogliosis, where terminally sick RML+LPS-treated mice exhibited significantly higher levels of astrogliosis across all brain regions compared to the RML-treated group (Figure 7, q-x). Furthermore, Ap staining results indicated similar levels of Ap deposition in both treatment groups, with only the Mb region of the RML-treated animals exhibiting Ap deposition (Figure 8, q-x).
In the case of terminally sick moPrPRes-treated mice, the treatment combination of moPrPRes and LPS resulted in a distinct vacuolation pattern. Terminally sick moPrPRes-treated mice exhibited a greater abundance of vacuolation across all brain regions compared to the moPrPRes+LPS treatment group. This difference was evident in the distribution patterns of vacuoles, with widespread vacuolation observed in terminally sick moPrPRes-treated mice, while the moPrPRes+LPS-treated group displayed a more limited distribution of vacuoles (Figure 4, i-p).
No accumulation of PrPSc was detected in either the moPrPRes-treated or the moPrPRes+LPS-treated terminally sick mice (Figure 6, i-p). Results from astrogliosis staining demonstrated a slightly more intense presence of astrocytes in the moPrPRes+LPS treatment group within the Cc, Th, and Mb compared to the moPrPRes treatment group (Figure 7, i-k and m-o). Conversely, the Cr region of the moPrPRes-treated mice exhibited more intense astrocyte accumulation compared to the moPrPRes+LPS treatment group (Figure 7, l and p). Importantly, neither of the moPrPRes-treated groups showed signs of Ap deposition in the brains of terminally sick mice (Figure 8, i-o).
Figure 9.
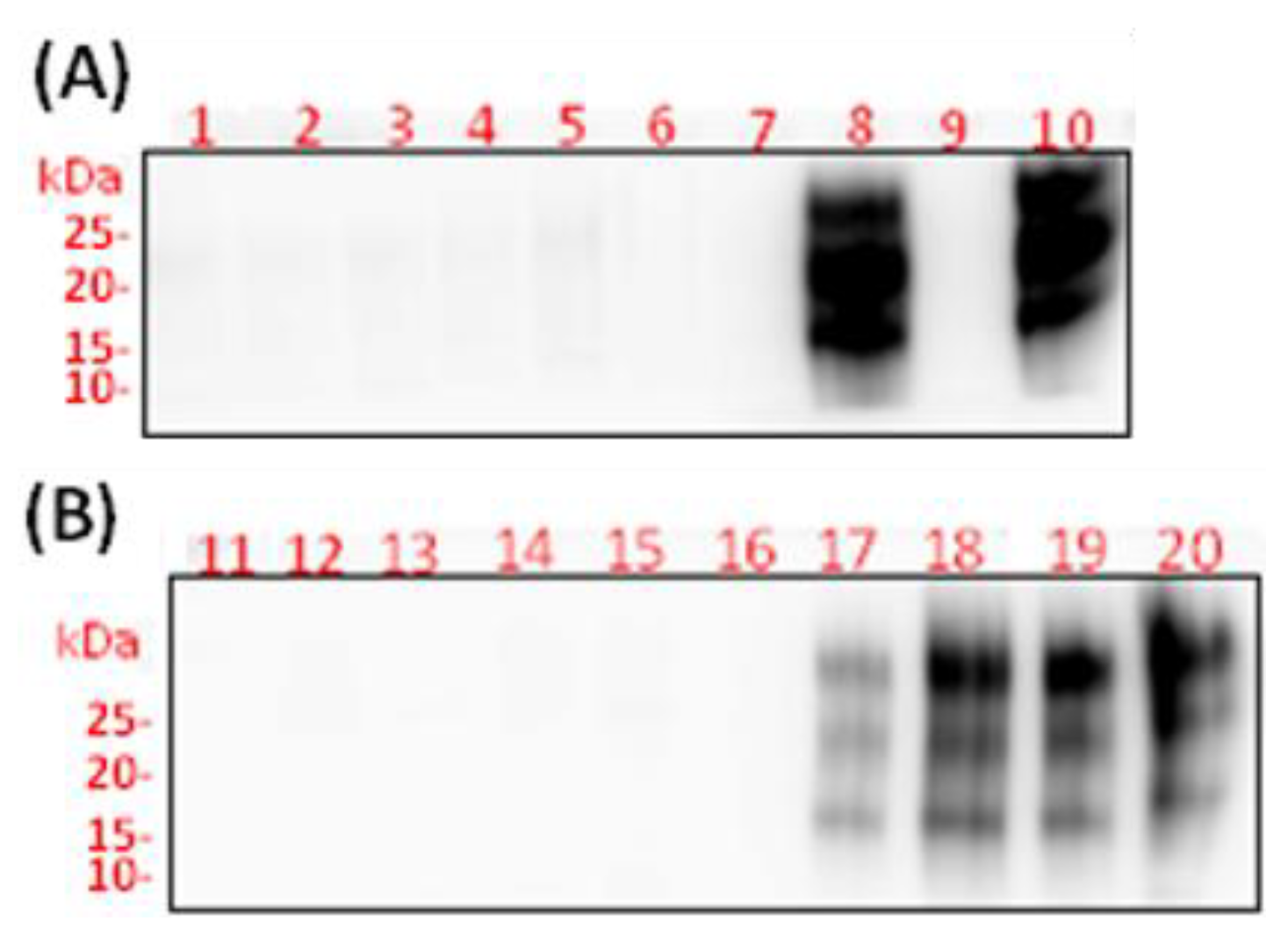
Western blot analyses of brain (A) and spleen (B) homogenates from FVB/N female mice inoculated subcutaneously with saline (negative control) (lanes 1 and 11), LPS from Escherichia coli 0111:B4 for 6 weeks (lanes 2 and 12), moPrPRes (lanes 3-4 and 13-14), moPrPRes + LPS (lanes 5-6 and 15-16), RML+LPS (lanes 7-8 and 17-18), and RML (positive control) (lanes 9-10 and 19-20). All samples were treated with 50 µg/mL proteinase K. The moPrPres- and RML-containing treatment groups (lanes 3-10 and 13-20) include samples from both 11 weeks (lanes 3, 5, 7, 9, 13, 15, 17, and 19) and terminally sick mice (lanes 4, 6, 8, 10, 14, 16, 18, and 20). Lane 0 contains the molecular weight ladder.
Figure 9.
Western blot analyses of brain (A) and spleen (B) homogenates from FVB/N female mice inoculated subcutaneously with saline (negative control) (lanes 1 and 11), LPS from Escherichia coli 0111:B4 for 6 weeks (lanes 2 and 12), moPrPRes (lanes 3-4 and 13-14), moPrPRes + LPS (lanes 5-6 and 15-16), RML+LPS (lanes 7-8 and 17-18), and RML (positive control) (lanes 9-10 and 19-20). All samples were treated with 50 µg/mL proteinase K. The moPrPres- and RML-containing treatment groups (lanes 3-10 and 13-20) include samples from both 11 weeks (lanes 3, 5, 7, 9, 13, 15, 17, and 19) and terminally sick mice (lanes 4, 6, 8, 10, 14, 16, 18, and 20). Lane 0 contains the molecular weight ladder.
2.3.6. Western Blot
Western blot analyses, along with PK treatment of brain homogenates and spleen samples, were conducted to assess the presence of disease-associated PrPRes at 11 wpi in both euthanized and terminally sick FVB/N female mice injected with LPS, moPrPRes, or RML (Figure 9). Our results indicated that both brain and spleen homogenates from saline-treated mice showed no bands corresponding to PrPRes (Figure 9, lanes 1 and 11). Interestingly, the positive RML-treated controls did not display any bands of PrPSc in the brain homogenates at 11 wpi (Figure 9A, lane 9).
Notably, RML-treated mice that were euthanized early at 11 wpi, despite showing no clinical signs of prion disease, exhibited detectable levels of PrPSc in their spleen homogenates (Figure 9B, lane 19). This finding was corroborated by the presence of detectable PrPSc levels in the spleen homogenates of terminally sick mice from the RML-treated group (Figure 9B, lane 20). The RML+LPS treated group exhibited similar results, with intense levels of PrPSc detected in the brain homogenates of terminally sick mice (Figure 9A, lane 10) and detectable levels of PrPSc in spleen homogenates at both 11 wpi and terminal illness (Figure 9B, lanes 17 and 18).
In contrast, all groups treated with LPS and/or moPrPRes displayed no detectable PrPSc in either brain or spleen homogenates at 11 wpi or at the terminal stage of the disease (Figure 9, lanes 2-7 and 12-16).
2.3.7. Scrapie Cell Assay
The L929 mouse fibroblast cell line was utilized to infect cells with brain homogenates from terminally sick mice across each treatment group, allowing for the visualization of prion-infected cells. Following the loading of cells onto ELISPOT plates, the samples were digested with proteinase K (PK) to facilitate the detection of PrPSc.
The results showed that all negative control wells, including those with uninfected saline brain homogenates and L929 cells, exhibited no positive signals. In contrast, the positive RML controls and the RML+LPS treatment group demonstrated a clear PrPSc-positive signal (Figure 10). Conversely, all other treatment groups that received LPS and/or moPrPRes displayed a PrPSc-negative signal, with values falling below the established cut-off threshold (Figure 10).
3. Discussion
3.1. Overview of Key Findings
This study makes three major contributions to our understanding of prion-like and inflammatory neurodegenerative processes. First, we show that a recombinant prion protein (moPrPRes) generated entirely in vitro by incubation briefly with LPS—without either seeding factors or serial protein misfolding cyclic amplification (sPMCA)—can provoke a distinctive neurodegenerative phenotype in wild-type FVB/N mice when administered subcutaneously. Second, we provide evidence that 6-week subcutaneous exposure to bacterial LPS, in the absence of a classical prion agent, is sufficient to induce Alzheimer’s-like spongiform brain pathology. Third, we demonstrate that the co-administration of LPS with the RML prion strain significantly accelerates disease onset, enhances neuropathological severity, and amplifies prion protein deposition, underscoring the powerful synergistic interplay between inflammatory triggers and classical prion agents.
Taken together, these findings highlight how bacterial factors and protein misfolding processes can converge or operate in parallel to drive neurodegenerative pathology. They also challenge the prevailing assumption that prion-related neurodegeneration is predicated solely on the accumulation of detectably protease-resistant prion protein (PrPSc) [1,2]. Instead, our data suggest multiple molecular routes—both prion-dependent and prion-independent—can converge upon a final common pathway of neuronal damage.
3.2. Infectivity and Pathogenic Potential of In Vitro-Generated moPrPRes
3.3. moPrPRes as a Novel Misfolded Protein Entity
A pivotal outcome of this research is that moPrPRes, produced through in vitro conversion using E. coli 0111:B4 LPS [5,6], can induce a pronounced spongiform encephalopathy in FVB/N mice without overt PrPSc accumulation. This finding expands upon prior demonstrations that exogenous cofactors, such as phospholipids or RNA, can foster prion protein misfolding in vitro [3,4]. However, in many cases, the resulting protease-resistant prion forms fail to cause disease in vivo unless further amplified by serial PMCA or supplemented with seeding factors [3,4]. By contrast, the moPrPRes described herein arose without additional amplification steps, indicating that a purely recombinant misfolded prion protein can be pathogenic in a wild-type host.
The neuropathological profile—featuring spongiform changes, significant astrogliosis (especially in the cerebellum), and a collection of clinical signs—resembles a prion-like disorder but deviates from classical scrapie in that PrPSc was undetectable by Western blot, immunohistochemistry, or scrapie cell assays [25]. This discrepancy strongly suggests that misfolded or partially aggregated conformers of PrP may be neurotoxic, even in the absence of large, protease-resistant deposits [3,4]. Alternatively, moPrPRes might invoke alternative neurodegenerative cascades, independent of the canonical prion replication pathway that yields abundant PrPSc.
3.4. Lack of Detectable PrPSc
The lack of classical protease-resistant prion aggregates in moPrPRes-treated mice raises fundamental questions about the centrality of PrPSc deposition to prion disease. It is widely accepted that the accumulation of PrPSc is the hallmark of prion disorders [1,2], yet increasing evidence indicates that smaller oligomeric or intermediate conformers may be the key mediators of neurotoxicity [3,4]. Our data suggest that moPrPRes can elicit pathological changes and clinical signs in the absence of overt PrPSc accumulation. This aligns with theories in other neurodegenerative proteinopathies (e.g., Alzheimer’s disease) that smaller, soluble aggregates, rather than large fibrillar deposits, may drive neuronal dysfunction.
3.5. Distinctions from RML-Derived Disease
Comparisons with RML (brain homogenate) treatments exposed substantial differences in incubation period, mortality rates, and neurodegeneration patterns. Whereas RML-inoculated mice typically succumbed by ~200–250 days post-infection (dpi), moPrPRes-treated animals exhibited significantly prolonged survival, with some remaining alive beyond 700 dpi. This disparity likely reflects the comparatively high infectivity of prion-containing brain homogenates [12] and the presence of pro-inflammatory or cofactor molecules that accelerate disease [4,12]. The relatively “pure” nature of moPrPRes, lacking these additional elements, may impede peripheral prion propagation and delay its ultimate neurotoxic effects. From a translational standpoint, these findings reinforce the notion that the composition of the inoculum—including inflammatory mediators and co-factors—critically influences disease onset and outcome [12,25].
4. LPS-Induced Neurodegeneration
4.1. Chronic LPS Infusion and Neuroinflammatory Damage
Equally striking is our discovery that a six-week subcutaneous infusion of LPS alone produces extensive brain spongiform vacuolation, prominent neuroinflammation, and ~40% mortality in wild-type FVB/N mice. Although acute or single-dose injections of LPS have long been known to provoke neuroinflammation [8,9,15,16,26], the robust Alzheimer’s-like pathology observed here underscores that sustained, low-dose endotoxin exposure can be a potent driver of de novo neurodegeneration in otherwise healthy animals.
These results accord with emerging data indicating that “metabolic endotoxemia” can catalyze systemic inflammation, impair blood–brain barrier integrity, and prime microglia for hyperactivation in a variety of models [27,28]. Chronic peripheral LPS exposure has been shown to upregulate TNF, IL-1β, MCP-1, and NF-κB p65, facilitating neuroinflammatory cascades that remain active for months [16]. Over time, this heightened inflammatory state may transition into progressive neuronal injury, reminiscent of what is observed in other protein misfolding diseases [18,29].
4.2. Metabolic Alterations and Obesity
An additional observation is that LPS-only mice developed significant weight gain after ~30 weeks of age—a result consistent with prior reports linking chronic endotoxin exposure to obesity, insulin resistance, and systemic inflammatory dysfunction [27,30]. Adipose tissue, particularly in the abdominal region, may act as a reservoir for pro-inflammatory mediators [27,28], potentially exacerbating neuroinflammation via endocrine or paracrine signaling pathways. Although unraveling the precise interplay between obesity, neurodegeneration, and inflammation lies beyond the scope of this article, our findings highlight the multifaceted consequences of chronic LPS exposure. Future investigations could clarify whether preventing metabolic derangements or targeting obesity-related inflammation might mitigate LPS-induced neuropathology.
5. Synergistic Impact of LPS and RML
5.1. Acceleration of Classical Prion Disease
The most rapid and severe disease manifested in the RML+LPS co-treatment group, characterized by a shorter incubation period, earlier onset of clinical signs, and enhanced pathology in the cerebral cortex, thalamus, midbrain, and cerebellum. Extensive PrPSc deposition, astrogliosis, and spongiform changes surpassed those seen in RML-only mice, implicating systemic inflammation as a catalyst that magnifies prion spread or replication [8,12,16]. Indeed, earlier research suggests that inflammatory states can alter the trafficking of prions through lymphoid tissues and facilitate their accumulation in the spleen and CNS [12,21,31]. In keeping with this notion, we detected PrPSc in the spleens of infected mice at 11 weeks post-infection (wpi), underscoring the role of lymphoreticular tissues as an early reservoir [11].
5.2. Contrasts with moPrPRes + LPS Co-Treatment
By comparison, LPS did not significantly exacerbate pathology or mortality in the moPrPRes group, which remained devoid of detectable PrPSc. This contrast underscores the complexity of prion biology and the influence of cofactors embedded in brain-derived scrapie isolates (i.e., RML) [3,4]. While chronic inflammation can heighten neuronal susceptibility, an “incomplete” or alternate prion conformer like moPrPRes may lack key features—such as seeding capacity or fully transmissible fibrillar structures—needed for replication and synergy with LPS. These findings speak to the importance of the prion strain’s molecular composition in shaping the disease course and the interactive role of immune factors [4,12].
5.3. Early Detection in the Spleen and Diagnostic Implications
Consistent with previous work demonstrating that spleen homogenates can reveal prion replication ahead of clinical onset [21,25,31], we found that RML-treated animals (with or without LPS) harbored detectable PrPSc in spleen samples as early as 11 wpi. This reaffirms the spleen as a pivotal site for prion replication and highlights its utility for presymptomatic diagnosis [31]. Notably, the co-presence of LPS amplified PrPSc accumulation in peripheral tissues, implicating systemic inflammatory responses in accelerating prion replication outside the CNS [12,16].
6. Mechanistic Insights and Future Directions
6.1. Unified Perspectives on Protein Misfolding and Inflammation
A central theme emerging from this study is that inflammation—whether triggered by bacterial products (LPS) or by prion-rich brain homogenates—can dramatically modulate neurodegenerative processes [12,16,32]. Chronic inflammation appears to promote microglial reactivity, alter blood–brain barrier integrity [33,34], and synergize with misfolded proteins to worsen neuronal damage. While the “protein-only” hypothesis posits that prion diseases are primarily driven by PrPSc conversion [1,3], our data underscore that infection and neurodegeneration are also shaped by systemic immune states, metabolic context, and the biochemical milieu in which misfolded proteins propagate [2,4,12].
6.2. Possible Roles of Oligomeric PrP and Alternative Neurotoxic Species
The moPrPRes variant may exist in oligomeric or partially resistant forms that elude conventional detection yet exert potent neurotoxicity—reminiscent of soluble aggregates implicated in Alzheimer’s disease [7,11]. Detailed biophysical characterization (e.g., using conformation-dependent immunoassays, electron microscopy, and advanced spectroscopic methods) could clarify whether moPrPRes forms these smaller, more toxic assemblies in vivo. Resolving these structural nuances is crucial for identifying new therapeutic targets, including strategies aimed at stabilizing harmless PrP conformations or preventing oligomer formation.
6.3. Therapeutic and Prophylactic Interventions
If bacterial endotoxin exposure can independently incite Alzheimer’s-like neuropathology and intensify classical prion disease, mitigating peripheral inflammation becomes a potentially valuable therapeutic approach. Anti-inflammatory drugs, or products that reduce endotoxin load, or interventions aimed at fortifying the intestinal barrier might reduce LPS influx, attenuate microglial hyperactivation, and slow disease progression [35,36,37]. Similarly, understanding how moPrPRes triggers neurotoxicity without robust PrPSc deposits could reveal strategies for counteracting harmful conformations of prion protein before they initiate irreversible neuronal damage.
7. Materials and Methods
7.1. Ethical Approval
This study was conducted in compliance with ethical standards and received approval from the University of Alberta Animal Care and Use Committee for Health Sciences Laboratory Animal Services. The procedures adhered to the guidelines established by the Canadian Council on Animal Care [22]. All aspects of animal care and use conformed to the University of Alberta's animal welfare laws, guidelines, and policies, ensuring that the welfare of the experimental animals was prioritized throughout the research.
7.2. Experimental Design and Animals
A total of ninety wild-type female FVB/N mice, sourced from Charles River Laboratories in Wilmington, USA, were utilized for this study. These mice were five weeks old at the start of the experiment and were randomly assigned to one of six treatment groups, each comprising fifteen mice. The treatment groups included: saline (negative control), LPS derived from Escherichia coli 0111:B4, moPrPRes (29-232) generated by incubation with E. coli 0111:B4 LPS (Sigma-Aldrich, St. Louis, USA), moPrPRes combined with LPS, RML (Rocky Mountain Laboratory) + LPS, and RML (positive control).
For the administration of treatments, bacterial LPS and saline were delivered subcutaneously using ALZET® osmotic mini pumps (ALZET, Cupertino, CA). These pumps were surgically implanted on the backs of the mice, allowing for continuous infusion at a rate of 0.11 µL/h over a period of six weeks. The dosage of bacterial LPS was set at 0.1 µg per gram of body weight. Concurrently, a single subcutaneous injection of either moPrPRes (29-232) at a dosage of 45 µg per mouse or RML, containing 107 ID50 units of scrapie prions, was administered at the time of pump implantation, according to the designated treatment group. The recombinant mouse prion protein (29-232) was provided by Dr. David Wishart’s laboratory at the University of Alberta and was injected in a total volume of 200 µL per mouse. For further details regarding the experimental design, readers can refer to the work by Hailemariam et al. [6].
7.3. Weight Measurement
To monitor the health and development of the mice throughout the study, body weight measurements were taken for each individual mouse in all treatment groups. This monitoring commenced at six weeks of age, immediately following the initiation of treatment. Weight measurements were conducted monthly to assess the overall health status of the mice. As the experiment progressed and mortality rates increased, the remaining mice from each treatment group were weighed again at 102 and 110 weeks of age, providing valuable data on the long-term effects of the treatments administered.
7.4. Mouse Recombinant Prion Protein
Lyophilized lipopolysaccharide (LPS) from Escherichia coli (Sigma-Aldrich, St. Louis, USA) was reconstituted with Milli-Q ddH₂O to achieve an initial working concentration of 5 mg/mL. This stock solution was then used to reconstitute lyophilized mouse recombinant prion protein (moPrP, 29-232) to approximately 0.5 mg/mL, resulting in a weight ratio of moPrP:LPS of 1:1 when mixed. Given that the average molecular weight of LPS is approximately 10 kDa, which is roughly half that of the prion protein, this weight ratio corresponds to a molar ratio of approximately 2:1. Due to the absence of a definitive molecular weight for LPS, weight (mg) ratios were utilized in this study to maintain consistency and facilitate comparison.
The prion conversion reactions were monitored using circular dichroism (CD) spectroscopy, performed in the far-UV region (190-260 nm) at 25 ºC in a 0.02 cm path-length quartz cell using an Olis DSM 17 spectropolarimeter. Five scans were averaged for each CD spectrum collected, with protein concentrations maintained at 0.5 mg/mL (25 µM). Reference spectra were acquired under the same conditions and were subtracted from the measured prion spectra prior to calculating molar ellipticity. Molar ellipticity values were determined using an average amino acid molecular weight of 113.64 g/mol. Additionally, the secondary structure content was analyzed with CDPro, employing the CONTINLL algorithm and the SP22X reference set. The endpoint for β-sheet conversion was defined when the helical content (as measured by CD) dropped to 15% and the β-sheet content exceeded 25%. The endpoint for fibril propagation was established when the helical content fell to 10% and the β-sheet content surpassed 30%.
To facilitate conversion, lyophilized moPrP was reconstituted to 0.5 mg/mL with the previously prepared LPS solution. Following this, LPS was removed from the LPS-converted prion solution using polymyxin B agarose resin (Sigma-Aldrich, St. Louis, USA). Five hundred µL of the resin was added to a 1.5 mL Eppendorf tube and equilibrated with three separate volumes of 500 µL ammonium bicarbonate buffer (100 mM, pH 8.0; Sigma-Aldrich, St. Louis, USA). Subsequently, 250 µL of the PrP sample was added and incubated at room temperature for 60 minutes, allowing any unbound LPS to attach to the resin. The resin was then pelleted by centrifugation at 850 g for 5 minutes, and the supernatant was discarded. This washing procedure was repeated three additional times, with the supernatant applied to freshly equilibrated resin. The final supernatant was analyzed using the Pyrochrome Limulus amebocyte lysate assay (Associates of Cape Cod Inc., East Falmouth, MA) to quantify any residual LPS contamination, while the secondary structure of the moPrPres was confirmed by CD spectroscopy.
7.5. Euthanasia
The euthanasia of the animals was conducted at two critical time points: 11 weeks post-infection (wpi) and at the terminal stage of the disease. Five mice from each treatment group were euthanized at 11 wpi, all exhibiting normal body weights and showing no clinical signs of prion disease or any abnormalities. The remaining ten mice per treatment group were euthanized at the terminal stage, characterized by clinical signs including kyphosis, ataxia, dysmetria, tremors, head tilt, tail rigidity, bradykinesia, proprioceptive deficits, stupor, loss of deep pain sensation, and significant weight loss.
7.6. Tissue Preparation
Prior to euthanasia, mice were anesthetized using isoflurane gas. Reflexes were assessed to ensure loss of pain sensation before proceeding with euthanasia through cardiac puncture to draw all blood. Following euthanasia, brain samples were collected immediately and sectioned in a sagittal orientation. The samples were fixed in 10% buffered formalin phosphate (Sigma-Aldrich, St. Louis, USA), using a fixation volume ten times greater than that of the tissue for a minimum of 48 hours at room temperature. After fixation, brain samples were rinsed with tap water for 20 minutes and subsequently placed in 15 mL tubes containing 70% ethanol (Commercial Alcohol, Winnipeg, Canada). The fixed brain samples were stored at 4 ºC until further processing for immunohistochemical (IHC) staining.
The samples underwent a series of IHC stainings to evaluate the pattern and distribution of PrPSc and amyloid plaque (Ap) deposition across various regions of the brain, along with assessments of astrogliosis and spongiform vacuolation.
7.7. Hematoxylin and Eosin Staining
Brain tissue samples were processed following the methodology described by Chishti et al. [23]. All brain tissues were mounted on adhesive-treated slides and embedded in paraffin (Formula “R” paraffin, Surgipath®, Leica Biosystems, Nussloch, Germany), then dried overnight at 37 ºC. The paraffin-embedded tissues were deparaffinized and rehydrated using a graded series of ethanol and deionized water. Slides were then incubated in filtered Mayer’s Hematoxylin (Fisher Scientific, Waltham, USA) and subsequently in eosin Y. After drying, images of each slide were captured using a digital slide scanner (NanoZoomer XR, Hamamatsu, SZK, Japan).
7.8. PrPSc Staining
Brain samples were processed for PrPSc staining following the protocol outlined by Bell et al. [24]. The procedure aimed to achieve maximum clearance of PrPC from the tissues. A series of incubations were performed using the following reagents: formic acid, 4 M guanidine thiocyanate, and 3% hydrogen peroxide (H2O2). Afterward, the samples were treated with a mouse monoclonal SAF83 antibody (1:500 dilution; Cayman Chemical, Michigan, USA), followed by incubation with streptavidin-peroxidase. The development of the staining was facilitated by the addition of diaminobenzidine (DAB) substrate, and the slides were counterstained with Mayer’s Hematoxylin (Fisher Scientific, Waltham, USA) to enhance visualization.
7.9. Amyloid Plaque Staining
The amyloid plaque staining was conducted as described by Chishti et al. [23]. Brain slices were incubated in thioflavine S (Sigma-Aldrich, St. Louis, USA), which is a fluorescent dye specific for amyloid plaques. Following incubation, the samples underwent treatment with alcohol to differentiate and dehydrate the plaques. After drying in the dark, the samples were examined for amyloid plaque (Ap) deposition using a Nikon microscope (Eclipse 90i; Nikon Instruments Inc., Melville, USA).
7.10. Astrogliosis Staining
Astrogliosis staining was performed using the methodology established by Chishti et al. [23]. Following initial processing, brain slides were treated with a glial fibrillary acidic protein (GFAP) antibody (Sigma-Aldrich, St. Louis, USA; BD Pharmingen, Mississauga, Canada) to label reactive astrocytes. The samples were then incubated with streptavidin-peroxidase (Invitrogen, Carlsbad, USA) for 16 minutes, followed by treatment with DAB (BD Pharmingen, Mississauga, Canada) for up to 20 minutes to develop the characteristic brown color indicative of GFAP presence. A counterstaining with Mayer’s Hematoxylin (Fisher Scientific, Waltham, USA) was also performed. Subsequently, brain slices were treated with xylene (Sigma-Aldrich, St. Louis, USA) for clearing. Finally, the slides were mounted with Cytoseal (Fisher Scientific, Waltham, USA) and left to dry at room temperature for 48 hours.
7.11. Western Blot Assay
Brain and spleen samples were prepared to create a 10% tissue homogenate in phosphate-buffered saline (PBS) (1X; Bio-RAD, Hercules, USA). The total protein concentration was quantified using a bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Waltham, USA), with 250 µg of protein obtained from brain samples and 400 µg from spleen samples. Each protein sample was then diluted in 250 µL of radioimmunoprecipitation assay buffer (RIPA; Sigma-Aldrich, St. Louis, USA), and 50 µg/mL of proteinase K (PK) was added to the homogenate.
To facilitate visualization, 2 µL of 0.02% bromophenol blue (Bio-RAD, Hercules, USA) was incorporated into the solution. The samples were briefly vortexed and allowed to digest with PK for one hour at 37 °C. The reaction was subsequently terminated by adding 25 µL of phenylmethylsulfonyl fluoride (PMSF) to achieve a final concentration of 5 mM (Sigma-Aldrich, St. Louis, USA). After cooling the samples at room temperature for 5 minutes, they were centrifuged at 20,000 g for 60 minutes at 4 °C. The supernatant was discarded, and 15 µL of 2X sample buffer (SB) was added to the resulting pellet, followed by boiling for 10 minutes to denature the proteins.
Each sample (20-25 µL) was loaded into the corresponding wells of NuPAGE Bis-Tris mini gels (Life Technologies, Carlsbad, USA). The samples were then subjected to electrophoresis using a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) apparatus (Bio-RAD, Hercules, USA) at 200 volts for 50 minutes. Chemiluminescent molecular weight standards (Precision Plus Protein™ WesternC Standards, Bio-RAD, Hercules, USA) were included in each gel for reference.
Membrane transfer was performed overnight using a wet Western blotting apparatus (Bio-RAD, Hercules, USA) at a voltage of 20 volts. Following the transfer, membranes were quickly rinsed in 1X TBS-Tween (0.5%; Sigma-Aldrich, St. Louis, USA) and incubated with the prion protein monoclonal Sha31 antibody (1:30,000; Bertin Pharma, Montigny le Bretonneux, France) in 1X TBS-Tween (0.5%) overnight at 4 °C. After washing, the membranes were incubated with a secondary goat anti-mouse horseradish peroxidase (HRP) conjugate antibody (1:10,000; Bio-RAD, Hercules, USA) in 1X TBS-Tween (0.1%) containing 5% skim milk (Carnation, Smucker Foods of Canada, Ontario, Canada).
Imaging of the blots was performed using Pierce® ECL Plus Western Blotting Substrate (Thermo Fisher Scientific, Waltham, USA) and captured with an ImageQuant LAS 4000 series imaging system (GE Healthcare Life Sciences, Quebec, Canada).
7.12. Scrapie Cell Assay
The standard scrapie cell assay methodology was adapted from Mahal et al. [25]. In this assay, L929 mouse fibroblast cells (ATCC, Manassas, USA) were exposed to prion samples prepared as brain homogenates in 1X phosphate-buffered saline (PBS) at a concentration of 10%. A series of control samples, including saline (negative control), Escherichia coli 0111
Lipopolysaccharide, moPrPres, moPrPres+LPS, RML+LPS, and RML (positive control), were prepared at a volume of 30 µL each and loaded into a 96-well tissue culture plate (Corning Costar, Tewksbury, USA). The samples underwent a dilution series ranging from 0.1% to 0.0001% and were tested in six replicates. To each well, 20 µL of a cell suspension containing approximately 5,000 L929 cells was added.
The plate was incubated at 37 °C for 3-5 minutes before adding 150 µL of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% horse serum (Sigma, St. Louis, USA). Following this, the plates were placed in a humidified incubator at 37 °C with 5% CO2 for a total of five days. After the incubation period, the cells were passaged twice (1:4 and 1:7 dilutions), with each passage followed by a 5-day incubation at 37 °C.
To prepare the ELISPOT assay, 96-well ELISPOT plates (Millipore, Billerica, USA) were activated by washing with 70% ethanol (60 µL for 3 minutes; Commercial Alcohol, Winnipeg, Canada) and rinsing three times with 1X TBS (Sigma-Aldrich, St. Louis, USA). Thirty µL of 1X TBS was added to the wells to keep the membranes wet, and 20,000 L929 cells were loaded into each well of the ELISPOT plate. The filtered cells on each plate were then subjected to vacuum and dried at 50 °C for one hour.
To digest the cells, 60 µL of RIPA lysis buffer containing 5 µg/mL proteinase K (PK; Invitrogen, Carlsbad, USA) was added to each well. The plates were incubated at 37 °C for 90 minutes, after which the lysis buffer was removed, and each well was washed three times with 1X TBS. Subsequently, 100 µL of 2 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, St. Louis, USA) prepared in TBS was added to each well, and the plates were incubated on a rocker (3D rotator, model 4631, Thermo Scientific, Waltham, USA) at room temperature for 10 minutes. After removing the PMSF, the wells were washed three times with 1X TBS.
Next, 100 µL of 3 M guanidine thiocyanate (GdnSCN; Fisher Scientific, Waltham, USA) was added to each well, followed by another 10-minute incubation on the rocker at room temperature. Afterward, GdnSCN was removed, and the wells were washed four times with 1X TBS. Subsequently, 100 µL of 5% skim milk (prepared in 1X TBS; Carnation®, Alberta, Canada) was added, and the plates were incubated at room temperature for 60 minutes. After removing the milk and washing three times with 1X TBS, the primary antibody (SAF83, 1:1,000 dilution in 1X TBS; Cayman Chemical, Michigan, USA) was added and incubated on the rocker for 120 minutes at room temperature. Following this, the SAF83 was washed away with three rinses of 1X TBS, and the secondary antibody (1:5,000 dilution, goat anti-mouse alkaline phosphatase conjugate; Bio-RAD, Hercules, USA) was added for a 90-minute incubation at room temperature on the rocker.
After removing the secondary antibody with three washes of 1X TBS, 60 µL of alkaline phosphatase buffer (100 mM Tris-HCl, 100 mM NaCl, 5 mM MgCl2·6H2O; Sigma-Aldrich, St. Louis, USA) was added to each well, followed by a 10-minute incubation on the rocker. The alkaline phosphatase buffer was then removed, and 60 µL of BCIP (5-bromo-4-chloro-3-indolyl-phosphate)/NBT (nitro blue tetrazolium) substrate solution (Promega, Madison, USA) was added, with subsequent incubation for 20 minutes. Following this, the BCIP/NBT solution was removed, and the wells were washed four times with distilled water. The plates were then allowed to dry overnight in the dark.
7.13. Statistics
Weight trends were analyzed using the MIXED procedure in SAS (version 9.3; SAS Institute, Cary, NC, USA). The statistical model utilized for the analysis was as follows:
Yijkl = μ + ti + pj + tpij + εijkl
In this equation, Yijkl represents the dependent variable, μ is the population mean, ti denotes the fixed effect of treatment, pj indicates the fixed effect of the period (age in weeks), tpij accounts for the interaction between treatment and period, and εijkl represents the residual error, which is assumed to follow a normal distribution. The degrees of freedom were calculated using the Kenward-Roger method. Results were presented through comparisons of least squares means, utilizing the SAS probability difference option. The significance level was set at a cut-off value of P < 0.05. Additionally, survival analysis was performed using PRISM software (Prism Software Corporation) to evaluate differences in survival rates across treatment groups.
8. Conclusions
Overall, this work provides multiple paradigm-shifting insights for prion biology and the broader field of neurodegenerative disorders:
1. Pathogenic moPrPRes without detectable PrPSc: We demonstrate that purely recombinant prion protein, converted in vitro with LPS but lacking classical PrPSc, can still induce neurodegeneration in wild-type mice.
2. LPS-driven Alzheimer’s-like pathology: Prolonged subcutaneous LPS exposure (6 weeks) alone is sufficient to cause widespread brain vacuolation, neuroinflammation, and a substantial mortality rate in otherwise healthy animals, suggesting that chronic bacterial endotoxin exposure may be a key driver of non-prion neurodegeneration.
3. Synergistic impact of LPS and RML: Co-administration of LPS with a classical prion strain (RML) dramatically exacerbates pathological outcomes, accelerating mortality and increasing spongiform changes and astrogliosis, thereby reinforcing the importance of inflammatory cofactors in prion diseases.
Ultimately, these insights underscore the multifactorial nature of prion diseases and highlight the importance of investigating similar inflammatory mechanisms in other protein misfolding syndromes. Further mechanistic work is warranted to dissect the molecular foundations of moPrPRes-induced neurodegeneration and to leverage these discoveries in the pursuit of novel strategies to prevent or mitigate prion-like disorders.
Author Contributions
Conceptualization, David Wishart and Burim Ametaj; Data curation, Grzegorz Zwierzchowski, Seyed Ali Goldansaz, Dagnachew Hailemariam and Burim Ametaj; Funding acquisition, David Wishart and Burim Ametaj; Investigation, Seyed Ali Goldansaz, Dagnachew Hailemariam, Elda Dervishi, David Wishart and Burim Ametaj; Methodology, David Wishart and Burim Ametaj; Project administration, David Wishart and Burim Ametaj; Resources, Roman Wójcik, David Wishart and Burim Ametaj; Validation, Seyed Ali Goldansaz, Dagnache Hailemariam, and Burim Ametaj; Writing – original draft, Seyed Ali Goldansaz, Dagnache Hailemariam, and Burim Ametaj; Writing – review & editing, Grzegorz Zwierzchowski, Seyed Ali Goldansaz, Dagnachew Hailemariam, Elda Dervishi, Roman Wójcik, David Wishart and Burim Ametaj. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the Alberta Livestock and Meat Agency Ltd. (ALMA) and the Alberta Prion Research Institute (APRI) under grant number 2010R068R, dated March 4, 2011.
Institutional Review Board Statement
All protocols implemented in this study adhered to the guidelines established by the Canadian Council on Animal Care and received approval from the Animal Care and Use Committee (Animal Use Protocol #660) at the University of Alberta's Health Sciences department.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated in this study are presented within this published article.
Acknowledgments
We would like to express our gratitude for the financial support of this research, which was provided by grants from the Alberta Livestock and Meat Agency and the Alberta Prion Research Institute, awarded to Drs. Burim N. Ametaj (Principal Investigator) and David Wishart (Co-Principal Investigator). We are particularly thankful to Dr. Suzanna M. Dunn for her assistance with laboratory procedures, and to Dr. Fozia Saleem for supplying the LPS-converted PrPRes used in this study. We also extend our appreciation to the staff at the Center for Prions and Protein Folding Diseases (CPPFD) at the University of Alberta for their exceptional care and monitoring of the experimental animals, as well as for conducting immunohistochemical (IHC) staining and scrapie cell assays (SCA). Special thanks are due to H. Gapeshina, Dr. J. Van der Merwe, Dr. Nathalie Daude, and J. Yang for their invaluable contributions. Finally, we acknowledge A. Abera for preparing the recombinant moPrP utilized in our experiments.
Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Soto, C. Prion hypothesis: the end of controversy? Trends Biochem. Sci. 2011, 36, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef]
- Supattapone, S. Cofactor molecules: essential partners for infectious prions. In [Book Chapter]; 2020; pp. 53–75. [Google Scholar] [CrossRef]
- Saleem, F.; Bjorndahl, T.C.; Ladner, C.L.; Perez-Pineiro, R.; Ametaj, B.N.; Wishart, D.S. Lipopolysaccharide induced conversion of recombinant prion protein. Prion 2014, 8, 221–232. [Google Scholar] [CrossRef]
- Hailemariam, D.; Goldansaz, S.A.; Daude, N.; Wishart, D.S.; Ametaj, B.N. Mice treated subcutaneously with mouse LPS-converted PrPres or LPS alone showed brain gene expression profiles characteristic of prion disease. Vet. Sci. 2021, 8, 200. [Google Scholar] [CrossRef]
- Bester, J.; Soma, P.; Kell, D.B.; Pretorius, E. Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget 2015, 6, 35284–35303. [Google Scholar] [CrossRef] [PubMed]
- Combrinck, M.I.; Perry, V.H.; Cunningham, C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 2002, 112, 7–11. [Google Scholar] [CrossRef]
- Liu, M.; Bing, G. Lipopolysaccharide animal models for Parkinson’s disease. Parkinsons Dis. 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Tiwari, P.C.; Pal, R. The potential role of neuroinflammation and transcription factors in Parkinson disease. Dialogues Clin. Neurosci. 2017, 19, 71–80. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef]
- Chianini, F.; Sisó, S.; Ricci, E.; Eaton, S.L.; Finlayson, J.; Pang, Y.; et al. Pathogenesis of scrapie in ARQ/ARQ sheep after subcutaneous infection: effect of lymphadenectomy and immune cell subset changes in relation to prion protein accumulation. Vet. Immunol. Immunopathol. 2013, 152, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.C.; Dorneles, G.P.; Santana Filho, P.C.; da Silva, I.M.; Schipper, L.L.; Postiga, I.A.L.; et al. Increased LPS levels coexist with systemic inflammation and result in monocyte activation in severe COVID-19 patients. Int. Immunopharmacol. 2021, 100, 108125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tang, B.; Wang, L.; Huo, Q.; Tan, S.; Misrani, A.; et al. Systemic LPS-induced microglial activation results in increased GABAergic tone: a mechanism of protection against neuroinflammation in the medial prefrontal cortex in mice. Brain Behav. Immun. 2022, 99, 53–69. [Google Scholar] [CrossRef]
- Johansson, S.; Bohman, S.; Radesäter, A.-C.; Öberg, C.; Luthman, J. Salmonella lipopolysaccharide (LPS) mediated neurodegeneration in hippocampal slice cultures. Neurotox. Res. 2005, 8, 207–220. [Google Scholar] [CrossRef]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.; et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef]
- Ametaj, B.N.; Sivaraman, S.; Dunn, S.M.; Zebeli, Q. Repeated oral administration of lipopolysaccharide from Escherichia coli 0111:B4 modulated humoral immune responses in periparturient dairy cows. Innate Immun. 2012, 18, 638–647. [Google Scholar] [CrossRef]
- Deng, X.; Li, M.; Ai, W.; He, L.; Lu, D.; Patrylo, P.R.; et al. Lipolysaccharide-induced neuroinflammation is associated with Alzheimer-like amyloidogenic axonal pathology and dendritic degeneration in rats. Adv. Alzheimer Dis. 2014, 3, 78–93. [Google Scholar] [CrossRef]
- Goldansaz, S.A.; Guo, A.C.; Sajed, T.; Steele, M.A.; Plastow, G.S.; Wishart, D.S. Livestock metabolomics and the livestock metabolome: a systematic review. PLoS One 2017, 12, e0177675. [Google Scholar] [CrossRef]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; et al. Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Hunter, N.; Foster, J.; Chong, A.; McCutcheon, S.; Parnham, D.; Eaton, S.; et al. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 2002, 83, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Canadian Council on Animal Care. Guide to the Care and Use of Experimental Animals; 1993; [cited 30 Sep 2021]. Available online: http://www.ccac.ca (accessed on 30 September 2021).
- Chishti, M.A.; Yang, D.-S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef]
- Bell, J.E.; Gentleman, S.M.; Ironside, J.W.; McCardle, L.; Lantos, P.L.; Doey, L.; et al. Prion protein immunocytochemistry – UK five centre consensus report. Neuropathol. Appl. Neurobiol. 1997, 23, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Mahal, S.P.; Demczyk, C.A.; Smith, E.W.; Klohn, P.-C.; Weissmann, C. Assaying prions in cell culture. In [Book Chapter]; 2008; pp. 49–68. [Google Scholar] [CrossRef]
- Xing, B.; Bachstetter, A.D.; Van Eldik, L.J. Microglial p38α MAPK is critical for LPS-induced neuron degeneration, through a mechanism involving TNFα. Mol. Neurodegener. 2011, 6, 84. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Lassenius, M.I.; Pietiläinen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjänen, J.; Forsblom, C.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef]
- Loffredo, L.; Ettorre, E.; Zicari, A.M.; Inghilleri, M.; Nocella, C.; Perri, L.; et al. Oxidative stress and gut-derived lipopolysaccharides in neurodegenerative disease: role of NOX2. Oxid. Med. Cell. Longev. 2020, 2020, 1–7. [Google Scholar] [CrossRef]
- Lawrence, C.B.; Brough, D.; Knight, E.M. Obese mice exhibit an altered behavioural and inflammatory response to lipopolysaccharide. Dis. Model Mech. 2012. [Google Scholar] [CrossRef]
- Urayama, A.; Morales, R.; Niehoff, M.L.; Banks, W.A.; Soto, C. Initial fate of prions upon peripheral infection: half-life, distribution, clearance, and tissue uptake. FASEB J. 2011, 25, 2792–2803. [Google Scholar] [CrossRef]
- Batista, C.R.A.; Gomes, G.F.; Candelario-Jalil, E.; Fiebich, B.L.; de Oliveira, A.C.P. Lipopolysaccharide-induced neuroinflammation as a bridge to understand neurodegeneration. Int. J. Mol. Sci. 2019, 20, 2293. [Google Scholar] [CrossRef]
- Van Amersfoort, E.S.; Van Berkel, T.J.C.; Kuiper, J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin. Microbiol. Rev. 2003, 16, 379–414. [Google Scholar] [CrossRef]
- Wolfs, T.G.A.M.; Derikx, J.P.M.; Hodin, C.M.I.M.; Vanderlocht, J.; Driessen, A.; de Bruïne, A.P.; et al. Localization of the lipopolysaccharide recognition complex in the human healthy and inflamed premature and adult gut. Inflamm. Bowel Dis. 2010, 16, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Cunningham, C.; Holmes, C. Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 2007, 7, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Heikenwalder, M. Pathogenesis of prion diseases: current status and future outlook. Nat. Rev. Microbiol. 2006, 4, 765–775. [Google Scholar] [CrossRef]
Figure 1.
Monthly weights of different treatment groups, including saline (negative control), bacterial lipopolysaccharide (LPS), mouse recombinant PrP proteinase-K resistant (moPrPres), moPrPres+LPS, RML+LPS, and RML (positive control). Lipopolysaccharide or saline were administered subcutaneously for 6 weeks through ALZET® osmotic mini pumps (ALZET, Cupertino, CA). A single injection of moPrPres or RML was administered at the time of mini pump implantation. Statistical comparisons indicate significant differences between LPS vs. RML (at 35, 39, 43, 48, 52, and 66 weeks), LPS vs. saline (48, 52, 56, 66, and 110 weeks; marked with a star), LPS vs. RML+LPS (35 and 39 weeks), LPS vs. moPrPres (43, 48, 52, 56, 61, 66, 70, and 74 weeks), LPS vs. moPrPres + LPS (39, 43, 48, 52, 56, 61, 66, 70, 74, 102, and 110 weeks), moPrPres vs. RML (35 weeks), moPrPres vs. RML+LPS (35 and 39 weeks), moPrPres+LPS vs. RML+LPS (39 weeks), moPrPres+LPS vs. saline (56 and 70 weeks), RML vs. RML+LPS (39 weeks), and RML+LPS vs. saline (39 weeks).
Figure 1.
Monthly weights of different treatment groups, including saline (negative control), bacterial lipopolysaccharide (LPS), mouse recombinant PrP proteinase-K resistant (moPrPres), moPrPres+LPS, RML+LPS, and RML (positive control). Lipopolysaccharide or saline were administered subcutaneously for 6 weeks through ALZET® osmotic mini pumps (ALZET, Cupertino, CA). A single injection of moPrPres or RML was administered at the time of mini pump implantation. Statistical comparisons indicate significant differences between LPS vs. RML (at 35, 39, 43, 48, 52, and 66 weeks), LPS vs. saline (48, 52, 56, 66, and 110 weeks; marked with a star), LPS vs. RML+LPS (35 and 39 weeks), LPS vs. moPrPres (43, 48, 52, 56, 61, 66, 70, and 74 weeks), LPS vs. moPrPres + LPS (39, 43, 48, 52, 56, 61, 66, 70, 74, 102, and 110 weeks), moPrPres vs. RML (35 weeks), moPrPres vs. RML+LPS (35 and 39 weeks), moPrPres+LPS vs. RML+LPS (39 weeks), moPrPres+LPS vs. saline (56 and 70 weeks), RML vs. RML+LPS (39 weeks), and RML+LPS vs. saline (39 weeks).
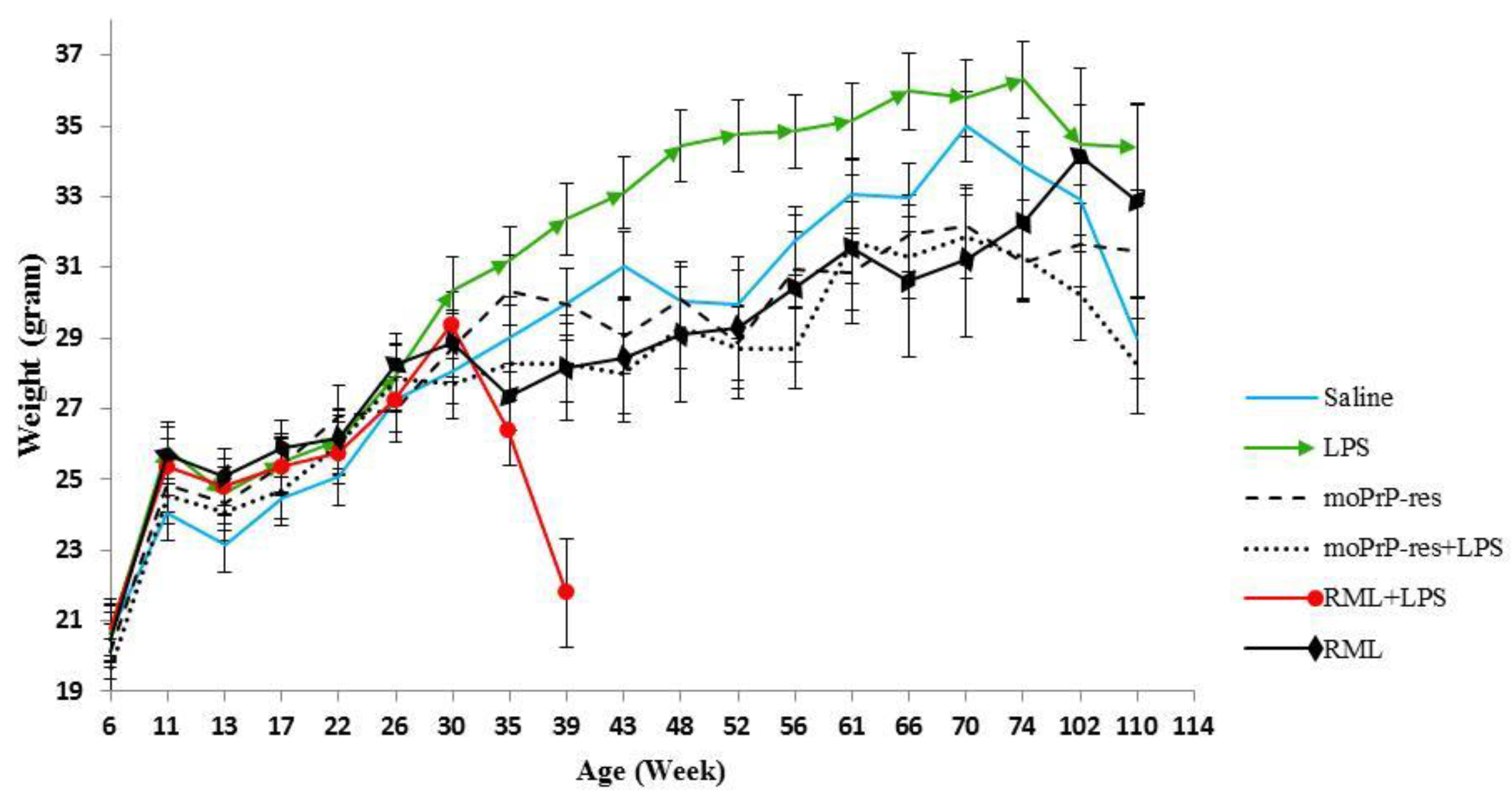
Figure 2.
Survival analysis of terminally sick FVB/N female mice. All mice were monitored for clinical signs of prion disease until 750 days post-inoculation and were subsequently euthanized or found dead after maintaining clinical signs for a minimum of 72 h. Negative controls (saline-treated animals) exhibited no clinical signs of prion disease, with all being found dead in the cage without prior symptoms. Most treated animals were euthanized or found dead with clinical signs resembling prion disease. Twenty percent of the RML positive controls survived until the termination of the experiment. The combination of RML with LPS caused earlier mortality (within 100–150 days), with all mice dead by 200 days. Sixty percent of the moPrPres-treated mice died before termination, with four showing clinical signs of neurodegeneration. The moPrPres+LPS treatment group had a mortality rate of 50%, with three mice showing clinical signs. Additionally, 40% of the LPS-treated animals exhibited clinical signs and were subsequently euthanized.
Figure 2.
Survival analysis of terminally sick FVB/N female mice. All mice were monitored for clinical signs of prion disease until 750 days post-inoculation and were subsequently euthanized or found dead after maintaining clinical signs for a minimum of 72 h. Negative controls (saline-treated animals) exhibited no clinical signs of prion disease, with all being found dead in the cage without prior symptoms. Most treated animals were euthanized or found dead with clinical signs resembling prion disease. Twenty percent of the RML positive controls survived until the termination of the experiment. The combination of RML with LPS caused earlier mortality (within 100–150 days), with all mice dead by 200 days. Sixty percent of the moPrPres-treated mice died before termination, with four showing clinical signs of neurodegeneration. The moPrPres+LPS treatment group had a mortality rate of 50%, with three mice showing clinical signs. Additionally, 40% of the LPS-treated animals exhibited clinical signs and were subsequently euthanized.
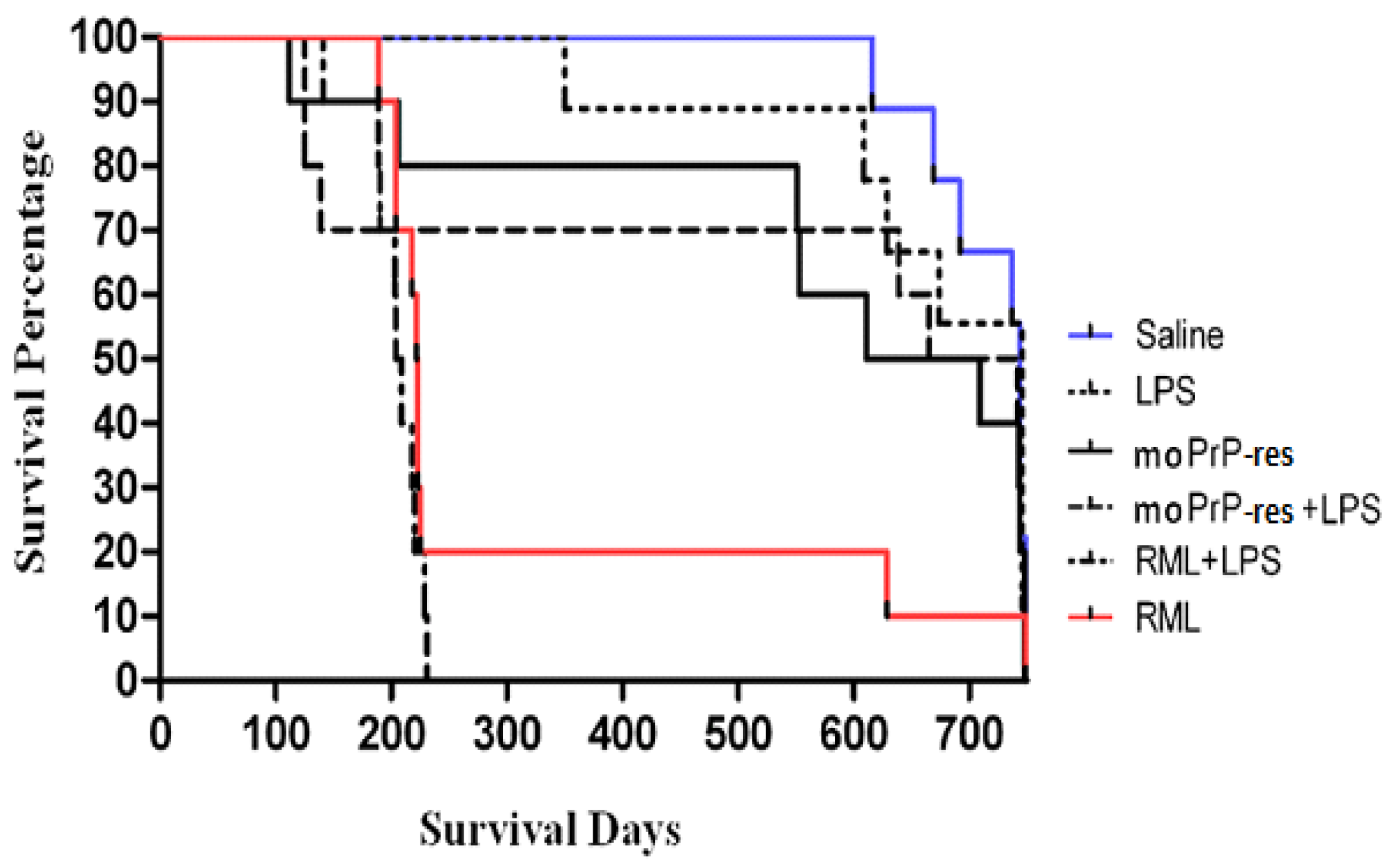
Figure 10.
Scrapie cell assay utilizing the L929 mouse fibroblast cell line and brain homogenates from terminally sick FVB/N female mice. Three brain homogenates from each of the terminally sick LPS, moPrPRes, moPrPRes+LPS, RML+LPS, and RML treatment groups were exposed to L929 cells in a dilution series of 0.1% to 0.0001%, with six replicates per dilution. The cut-off value was set at 150 spots/20,000 cells.
Figure 10.
Scrapie cell assay utilizing the L929 mouse fibroblast cell line and brain homogenates from terminally sick FVB/N female mice. Three brain homogenates from each of the terminally sick LPS, moPrPRes, moPrPRes+LPS, RML+LPS, and RML treatment groups were exposed to L929 cells in a dilution series of 0.1% to 0.0001%, with six replicates per dilution. The cut-off value was set at 150 spots/20,000 cells.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



