Submitted:
02 February 2026
Posted:
05 February 2026
You are already at the latest version
Abstract
Whole-genome sequencing (WGS) is a powerful tool for uncovering genome-wide variation, identifying selection signatures, and guiding genetic improvement in livestock. Royal White (RW) and White Dorper (WD) sheep are economically important meat-type hair breeds in the U.S., yet their genomic architecture remains poorly characterized. In this study, WGS was performed on 20 ewe sheep (n = 11 RW, n = 9 WD) to identify and annotate SNPs and small insertions and deletions (indels). Functional annotation, gene enrichment, population structure, and selective sweep analysis were also performed. Selective sweep analysis was conducted by integrating the fixation index (FST), nucleotide diversity (π), and Tajima’s D to identify candidate regions under putative recent positive selection. A total of 21,957,139 SNPs and 2,866,600 indels were identified in RW, and 18,641,789 SNPs and 2,397,368 indels in WD. In RW sheep, candidate genes under selection were associated with health and parasite resistance (NRXN1, HERC6, TGFB2) and growth traits (JADE2). In WD sheep, selective sweep regions included genes linked to immune response and parasite resistance (TRIM14), body weight (PLXDC2), and reproduction (STPG3). These findings were supported by sheep-specific quantitative trait loci (QTL) annotations and SNP–trait associations. This study provides the first WGS-based genomic comparison between RW and WD sheep, establishing a foundation for future genetic improvement, including targeted selection for enhanced immune fitness, disease resistance, and other economically important traits in these breeds.
Keywords:
sheep
; ovine
; whole genome sequencing
; royal white
; white dorper
; SNP
; indel
; selective sweep analysis
; traits
1. Introduction
The domestic sheep (Ovis aries) is a globally important livestock species, contributing to food, fiber, and income security through the production of meat, wool, milk, and hides. According to statistical summaries from the Food and Agriculture Organization (FAO) of the United Nations for 2024, the global sheep population reached approximately 1.5 billion head worldwide [1]. In the U.S., the national flock totaled 5 million sheep and lambs as of January 1, 2025, including 3.7 million breeding sheep and 1.4 million market lambs [2]. Among U.S. sheep breeds, Royal White (RW) and White Dorper (WD) are prominent meat-type hair sheep valued for efficient meat production and adaptability, and they are also increasing in popularity in the U.S. due to their climate adaptability and lack of shearing requirements. Royal White® Sheep is a U.S.-developed composite breed created by Bill Hoag in the early 2000s, through the crossbreeding of Dorper and St. Croix sheep. The breed was developed to combine desirable traits such as carcass quality, parasite resistance, a clean-shedding hair coat, and adaptability to diverse production environments [3]. White Dorper is a South African meat-type hair sheep developed through strategic crossbreeding beginning in the 1930s. The breed originated from Dorset Horn rams imported from Australia and crossed with Blackhead Persian ewes, with later contributions from Van Rooy sheep. White Dorper shares identical breed standards with Blackhead Dorper, differing only in coat color and pigmentation [4]. Today, the breed is valued for its rapid growth, high fertility, adaptability, and broad use in arid production systems [5].
Given the RW and WD commercial importance, understanding the genomic architecture of these breeds is essential. Despite the growing economic relevance of U.S. meat production systems, genomic studies on RW and WD sheep remain limited. To date, no published studies have comprehensively characterized RW sheep and U.S. populations of WD sheep at the whole-genome level, leaving a critical gap in our understanding of their genetic architecture and selection history. While WD has been evaluated genomically in regions such as South Africa and Hungary using SNP chips [6,7], few genomic studies have focused on U.S. populations, limiting our understanding of how this breed adapts and performs under American production conditions. This gap is particularly relevant given that environmental pressures and selection objectives may differ across geographic regions, potentially shaping distinct genomic signatures. To address this gap, whole-genome sequencing (WGS) provides a powerful approach for capturing genome-wide variation, offering deeper insights into the genetic basis of breed adaptation, performance, and selection under specific production environments.
Whole-genome sequencing has substantially advanced livestock genomics by enabling comprehensive characterization of genetic variation across the entire genome. Unlike marker-based approaches, WGS captures both common and rare genetic variants at single-base resolution, including single nucleotide polymorphisms (SNPs) and small insertions and deletions (indels). These variants provide valuable insights into genetic diversity, population structure, breed evolution, and the genetic basis of traits under selection [8,9]. In livestock research, WGS facilitates the identification of putatively functional mutations and signatures of selection, offering a powerful framework for improving animal breeding, conservation, and adaptation strategies. Applying WGS to under-characterized breeds such as RW and U.S.-based WD sheep enables the discovery of breed-specific genomic features that may contribute to performance and environmental resilience. By applying WGS to both RW and WD sheep from the same flock, this study aims to uncover breed-specific genomic features and provide foundational insights for future breeding and conservation efforts.
The objectives of this study were to (1) identify and compare genome-wide genetic variants (including SNPs and indels) in RW and WD sheep using WGS, (2) annotate the functional impact of these variants and explore enriched biological pathways through Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, and (3) detect regions of the genome putatively under recent selection in each breed and identify candidate genes associated with key traits.
2. Materials and Methods
2.1. Sample Collection, DNA Extraction, Library Preparation, and Sequencing
All animal samples were obtained and experiments conducted according to the Virginia Polytechnic Institute and State University Institutional Animal Care and Use Committee (IACUC) #17-233 and Institutional Biosafety Committee (IBC) #20-067. A total of 20 ewe sheep, RW (Royal White; ) and WD (White Dorper; ), were used in this study. All animals originated from the same privately owned flock in the Southern U.S., minimizing environmental variation. The average age was (mean ± SE) years for RW and (mean ± SE) years for WD. For this study, a single blood sample was collected from each of the 20 animals. Whole blood (approximately 4 mL) was collected via jugular venipuncture into Ethylenediaminetetraacetic acid (EDTA) tubes under approved IACUC protocols. Samples were shipped overnight on ice, processed the next day, and peripheral blood mononuclear cells (PBMC) were isolated using density gradient centrifugation. Isolated PBMCs were washed, cryopreserved in CryoStor CS10, stored at –80°C for 24 h, and then transferred to liquid nitrogen until DNA extraction. For DNA extraction, up to thawed PBMCs were centrifuged at 300 for 6 minutes to obtain a cell pellet, which was resuspended in 200 µL phosphate-buffered saline (PBS) and mixed with 20 µL proteinase K. After the addition of 200 µL buffer AL (without ethanol; Qiagen; lysis buffer), samples were incubated at 56 °C for 10 minutes, followed by the addition of 200 µL of 100% ethanol. The mixture was then transferred to a spin column, washed with wash buffers AW1 (Qiagen, first wash buffer) and AW2 (Qiagen, second wash buffer), and DNA was eluted in 100 µL buffer AE (Qiagen, elution buffer). The concentration and purity of DNA were assessed using a NanoDrop ND-100 spectrophotometer. Libraries were prepared using the TruSeq DNA PCR-Free Library Preparation Kit (Illumina) following the manufacturer’s instructions. Libraries were sequenced on the Illumina NovaSeq 6000 platform with a paired-end 151 bp read length using the NovaSeq FASTQ workflow.
2.2. Alignment and Variant Calling
Raw reads were processed using Trimmomatic (version 0.39) [10], where adapter sequences were removed, low-quality bases (quality score < 30) were trimmed using a sliding window approach, and reads shorter than 50 bp were discarded. High-quality paired-end reads were aligned to the Ovis aries reference genome (ARS-UI_Ramb_v2.0) using BWA-MEM (version 0.7.18) [11] with default parameters. Aligned reads were first processed with GATK (version 4.6.1) [12] MarkDuplicates to identify and mark PCR duplicates. The resulting deduplicated BAM files were used for variant calling with GATK HaplotypeCaller in GVCF mode. Variants were initially called on a per-sample basis, and individual GVCF files were then combined by breed, RW and WD, using GATK CombineGVCFs. Joint genotyping was performed within each breed using GATK GenotypeGVCFs to produce breed-specific multi-sample VCF files. To obtain high-confidence variants, breed-specific VCF files were filtered using BCFtools (version 1.21) [13]. Variants were retained if they passed the following criteria: QUAL > 30, depth (DP) > 8, Fisher strand bias (FS) < 60.0, mapping quality (MQ) > 40.0, mapping quality rank sum (MQRankSum) >−12.5, quality by depth (QD) ≥ 2.0, read position rank sum (ReadPosRankSum) >−8.0, and strand odds ratio (SOR) ≤ 3.0. Filtered variants were compared against the Ensembl variation database (Ovis_aries_rambouillet; version 113; file date: 2024-08-29) to distinguish known and novel variants. The reference database contained a total of 89,897,984 variants, including 83,083,034 SNPs and 6,763,747 indels. Venn diagrams illustrating shared and unique variants between breeds were generated using the VennDiagram R package (version 1.7.3) [14]. The indel length distribution was visualized using a histogram generated with ggplot2 (version 3.5.1).
2.3. Functional Annotation and Gene Enrichment Analysis
Functional annotation of SNP and indel variant files was performed using SnpEff (version 5.2c) [15] with the Oar_ARS_UI_Ramb_v2_0 database. Annotated VCF files were generated for each breed and variant type. Following annotation, variants were filtered to retain those with predicted functional effects classified as HIGH or MODERATE impact by SnpEff. Genes affected by these variants were extracted from the annotated VCF files. To focus on genes with stronger predicted functional burden, only genes harboring at least five HIGH or MODERATE impact variants were retained for downstream analysis. The resulting gene lists were submitted to the Database for Annotation, Visualization and Integrated Discovery (DAVID, version 6.8) [16] for functional enrichment analysis. Gene ontology enrichment analysis focused on the biological process, cellular component, and molecular function categories, as well as KEGG pathways. The top 10 enriched terms from each GO category and from KEGG pathways, ranked by false discovery rate (FDR), were visualized in R using the ggplot2 package (version 3.5.1) based on their minus log10(FDR) values.
2.4. Population Structure Analysis
Prior to population structure analysis, variants were filtered to remove those with a call rate below 80% and a minor allele frequency (MAF) less than 0.05. Missing genotype values were imputed using Beagle (version 5.4) [17]. The filtered and imputed variant datasets for both RW and WD breeds were converted to PLINK binary format using PLINK2 (version 2.0) [18]. Population structure was assessed using principal component analysis (PCA) based on the top ten principal components. Principal component analysis was performed in PLINK2 using genotype data with standard allele-frequency–based scaling to account for differences in allele frequencies between breeds. The first two components were visualized using ggplot2 (version 3.5.1) in R (version 4.3.0) [19], with breed-specific coloring and ellipses to illustrate group separation. In addition to PCA, discriminant analysis of principal components (DAPC) [20] was conducted using the adegenet (version 2.1.11) package in R to further investigate breed-specific genetic differentiation. The first discriminant function was visualized as a density plot using ggplot2, with coloring consistent with PCA plots to enhance visual comparability.
2.5. Selective Sweep Analysis
Selective sweep regions were identified by integrating the population differentiation fixation index (FST), nucleotide diversity (), and Tajima’s D metrics. Window-based FST values were calculated using VCFtools (version 0.1.16) [21] with a sliding window of 50 kb and a step of 10 kb. Breed-specific nucleotide diversity () and Tajima’s D were computed separately for RW and WD using the same window parameters. The top 10% of genomic windows based on FST were selected as candidate regions of differentiation. Within these, regions with reduced diversity in one breed relative to the other were identified by calculating the natural log ratio of values [] and selecting the top and bottom 10% as WD- and RW-specific sweep candidates, respectively. To strengthen the evidence for putative recent selective sweeps, an additional filter was applied based on Tajima’s D. For each breed, only candidate regions with negative breed-specific Tajima’s D values were retained, indicating an excess of low-frequency alleles consistent with recent positive selection. Genomic positions in these regions were annotated with gene information. Genotypic identifiers were retrieved by cross-referencing positions with the Ensembl Ovis aries Rambouillet variation database (version 113). Trait associations were then incorporated by mapping SNP IDs to the sheep quantitative trait locus database (QTLdb; release 55; file date: 2024-12-23), enabling the identification of candidate genes linked to economically important traits.
3. Results
3.1. Read Quality, Mapping, and Depth Coverage
Whole-genome sequencing generated approximately 843 million raw paired-end reads across 11 RW sheep and 625 million raw paired-end reads across 9 WD sheep. After trimming, an average of 94.98% of RW reads and 92.95% of WD reads were retained. The average mapping rate for both breeds was consistently high at 99.93%, indicating excellent alignment quality. Royal White samples showed an average sequencing depth of 7.19× (ranging from 5.34× to 10.00×), while WD samples had an average depth of 6.33× (ranging from 5.01× to 7.41×) (Table 1).
3.2. Variant Calling
Variants were identified through within-breed joint genotyping using GATK and filtered to retain high-confidence SNPs and indels. A total of 26,555,583 SNPs and 3,703,099 indels were initially identified in RW, and 24,792,950 SNPs and 3,396,380 indels in WD. After quality filtering, 21,957,139 SNPs and 2,866,600 indels were retained in RW, and 18,641,789 SNPs and 2,397,368 indels in WD. The transition-to-transversion (Ts/Tv) ratio was 2.30 in RW and 2.16 in WD. The heterozygous-to-homozygous (Het/Hom) genotype ratio across called variants was 0.999 (SNPs) and 0.998 (indels) in RW and 0.998 (SNPs) and 0.992 (indels) in WD. Annotation against the Ensembl Ovis aries variation database (release 113) revealed that 77.24% of SNPs and 21.83% of indels in RW were known, while 22.76% and 78.17%, respectively, were novel. Similarly, 77.57% of SNPs and 21.87% of indels in WD were known, with 22.43% and 78.13% being novel (Table 2). Venn diagram analysis showed that RW and WD shared 13,498,534 SNPs and 1,350,346 indels, while 8,458,605 SNPs and 1,516,254 indels were unique to RW, and 5,143,255 SNPs and 1,047,022 indels were unique to WD (Figure 1). The length distribution of indels was examined to characterize insertion and deletion patterns in both breeds. As shown in Figure 2, the majority of indels in RW and WD sheep were short, with sizes concentrated between bp (deletions) and bp (insertions). One base-pair indels were the most frequent in both breeds, followed by two base-pair changes. The overall distribution was symmetric around 0 bp, with a peak at bp for insertions and bp for deletions. These results indicate that short indels are predominant in both populations.
3.2.1. Functional Annotation and Gene Enrichment Analysis
A comprehensive functional annotation of SNP and indel variant files was performed separately for RW and WD sheep using SnpEff to evaluate the potential biological effects of genomic variation within each breed. For SNPs, both breeds exhibited a high proportion of variants in non-coding regions, including introns (27.47M in RW; 23.39M in WD), intergenic regions (11.97M in RW; 10.14M in WD), downstream gene regions (2.33M in RW; 1.98M in WD), and upstream gene regions (2.31M in RW; 1.95M in WD). Functionally important categories such as missense variants were also prevalent (142,906 in RW; 124,264 in WD), suggesting protein-altering potential (Table 3). Additional variants were found in synonymous, splice region, and UTR regions. For indels, the most abundant categories included intron variants (4.29M in RW; 3.53M in WD) and intergenic variants (1.74M in RW; 1.42M in WD), followed by downstream and upstream gene variants. High-impact functional classes such as frameshift variants (10,427 in RW; 9,510 in WD), disruptive in-frame insertions/deletions, splice site variants, and stop-gainedd variants were also detected (Table 4). These findings indicate that both breeds harbor a substantial number of variants with the potential to affect gene function and regulation.
Gene ontology and KEGG enrichment analyses of genes harboring HIGH and MODERATE impact variants revealed broadly consistent functional categories and pathways in both RW and WD sheep (Figure 3 and Figure 4). In KEGG, both breeds were strongly enriched for ABC transporters (oas02010), ECM–receptor interaction (oas04512), graft-versus-host disease (oas05332), complement and coagulation cascades (oas04610), cytoskeleton in muscle cells (oas04820), amoebiasis (oas05146), and the Fanconi anemia pathway (oas03460), indicating potential roles in transmembrane transport, cell–matrix signaling, immune regulation, structural integrity, host–pathogen interactions, and DNA damage repair. Royal White-specific enrichment included retinol metabolism (oas00830) and linoleic acid metabolism (oas00591), suggesting potential breed-specific adaptations in vitamin A utilization and fatty acid processing that may influence growth and health. White Dorper-specific enrichment included Staphylococcus aureus infection (oas05150) and motor proteins (oas04814), pointing to putative differences in pathogen defense mechanisms, intracellular transport, and cytoskeletal regulation.

Table 4.
Summary of indel functional annotation categories in Royal White and White Dorper breeds based on SnpEff.
Table 4.
Summary of indel functional annotation categories in Royal White and White Dorper breeds based on SnpEff.
| Variant Type | Royal White Indels | White Dorper Indels |
|---|---|---|
| 3’ UTR truncation | 1 | 1 |
| 3’ UTR variant | 67,521 | 55,407 |
| 5’ UTR truncation | 1 | 3 |
| 5’ UTR variant | 21,424 | 17,480 |
| Bidirectional gene fusion | 78 | 68 |
| Conservative inframe deletion | 820 | 617 |
| Conservative inframe insertion | 654 | 479 |
| Disruptive inframe deletion | 1,473 | 1,098 |
| Disruptive inframe insertion | 671 | 509 |
| Downstream gene variant | 386,904 | 318,194 |
| Exon loss variant | 5 | 7 |
| Frameshift variant | 10,427 | 9,510 |
| Gene fusion | 59 | 30 |
| Intergenic region | 1,744,317 | 1,418,580 |
| Intragenic variant | 920 | 793 |
| Intron variant | 4,292,930 | 3,529,191 |
| Non-coding transcript exon variant | 22,460 | 19,116 |
| Non-coding transcript variant | 304 | 235 |
| Splice acceptor variant | 1,339 | 1,106 |
| Splice donor variant | 977 | 759 |
| Splice region variant | 12,640 | 10,121 |
| Start lost | 86 | 54 |
| Start retained variant | 24 | 17 |
| Stop gained | 135 | 127 |
| Stop lost | 64 | 56 |
| Stop retained variant | 10 | 10 |
| Transcript ablation | 1 | 2 |
| Upstream gene variant | 380,196 | 309,740 |
Figure 3.
The top 10 enriched terms from each functional category, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene Ontology (GO) molecular function, cellular component, and biological process, are shown, ranked by false discovery rate (FDR). Bars represent –log10(FDR) values. Enrichment analyses were based on genes carrying at least five SNPs or indels with HIGH or MODERATE impact.
Figure 3.
The top 10 enriched terms from each functional category, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene Ontology (GO) molecular function, cellular component, and biological process, are shown, ranked by false discovery rate (FDR). Bars represent –log10(FDR) values. Enrichment analyses were based on genes carrying at least five SNPs or indels with HIGH or MODERATE impact.
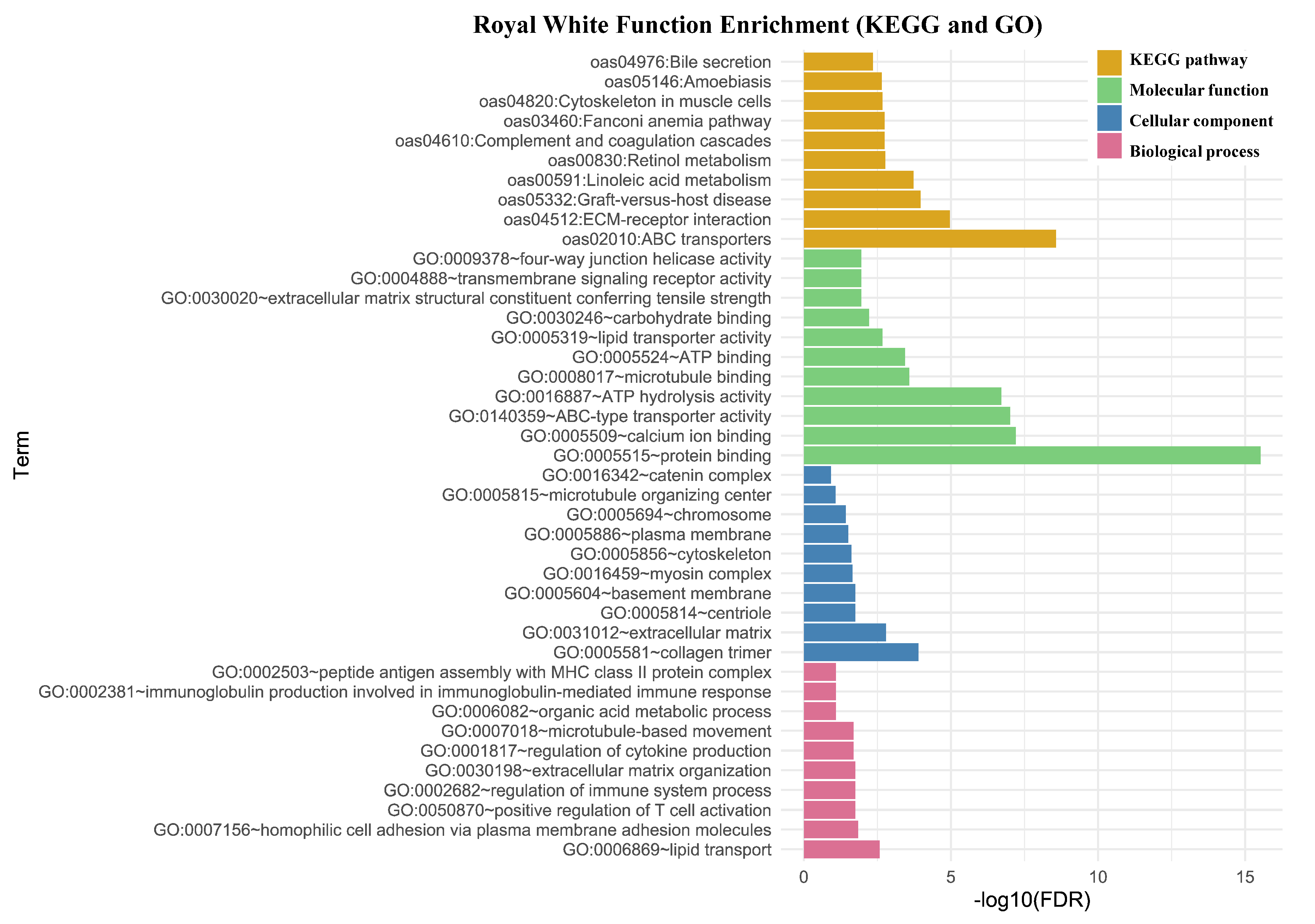
Figure 4.
The top 10 enriched terms from each functional category, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene Ontology (GO) molecular function, cellular component, and biological process, are shown, ranked by false discovery rate (FDR). Bars represent –log10(FDR) values. Enrichment analyses were based on genes carrying at least five SNPs or indels with HIGH or MODERATE impact.
Figure 4.
The top 10 enriched terms from each functional category, including Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway, Gene Ontology (GO) molecular function, cellular component, and biological process, are shown, ranked by false discovery rate (FDR). Bars represent –log10(FDR) values. Enrichment analyses were based on genes carrying at least five SNPs or indels with HIGH or MODERATE impact.
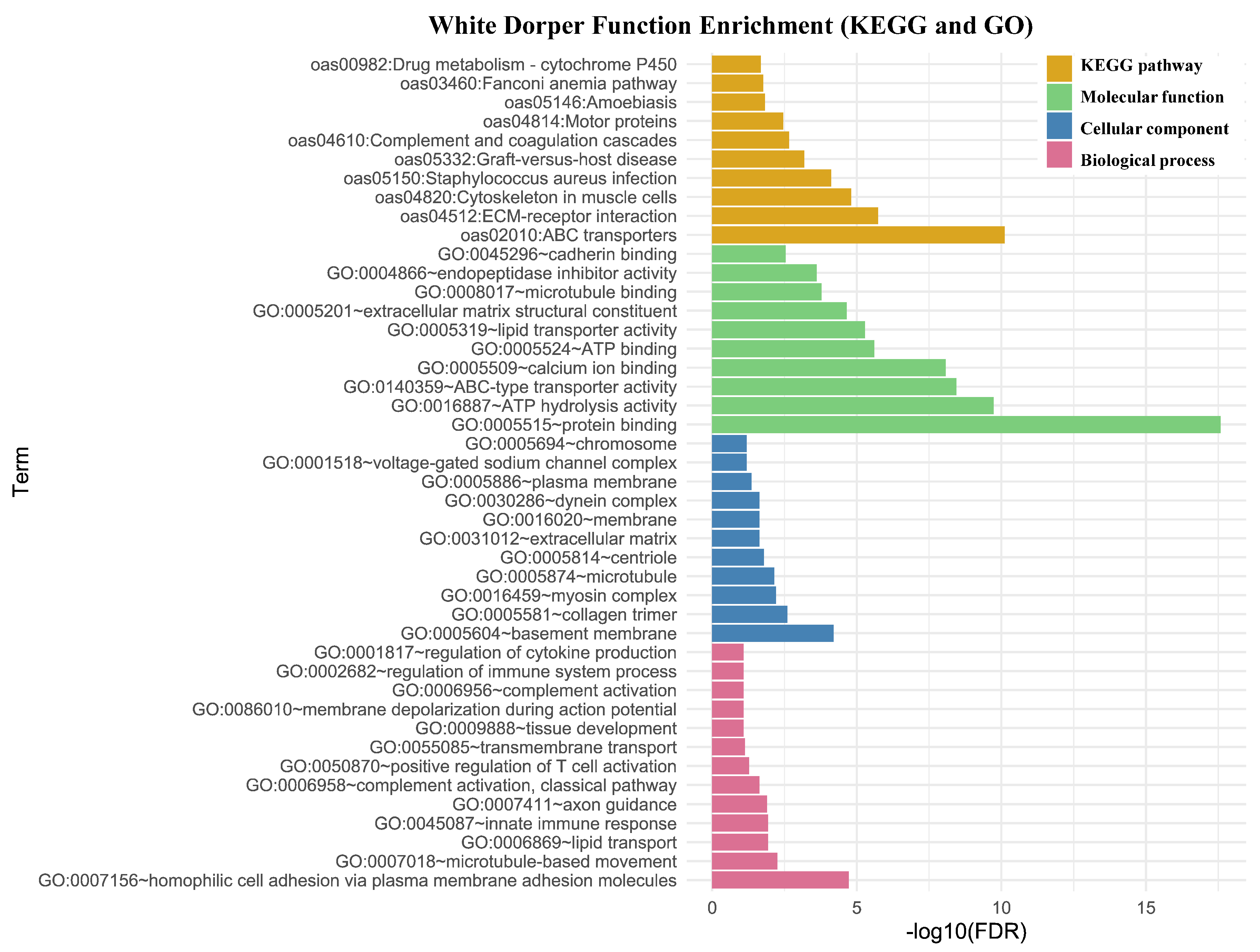
At the molecular function level, both breeds were significantly enriched for protein binding (GO:0005515), calcium ion binding (GO:0005509), ATP binding (GO:0005524), ATP hydrolysis activity (GO:0016887), ABC-type transporter activity (GO:0140359), lipid transporter activity (GO:0005319), microtubule binding (GO:0008017), and extracellular matrix structural constituent (GO:0005201 in WD; GO:0030020 in RW), suggesting potential common roles in energy metabolism, membrane transport, cytoskeletal interactions, and structural integrity. Royal White-specific terms included four-way junction helicase activity (GO:0009378), transmembrane signaling receptor activity (GO:0004888), and carbohydrate binding (GO:0030246), indicating possible breed-specific differences in DNA repair, signal transduction, and carbohydrate recognition. White Dorper-specific enrichment included cadherin binding (GO:0045296) and endopeptidase inhibitor activity (GO:0004866), pointing to potential variation in cell–cell adhesion and proteolysis regulation.
In the cellular component category, enrichment for collagen trimer (GO:0005581), myosin complex (GO:0016459), plasma membrane (GO:0005886), and extracellular matrix (GO:0031012) was observed in both breeds, reflecting structural and signaling roles commonly affected by coding variants. Notably, RW showed unique enrichment for the catenin complex (GO:0016342), microtubule organizing center (GO:0005815), and cytoskeleton (GO:0005856), suggesting enhanced roles in cytoskeletal organization and cell–cell adhesion. White Dorper showed additional terms like dynein complex (GO:0030286) and microtubule (GO:0005874), indicating potential differences in microtubule-based transport and structural dynamics.
Biological process terms were also partially shared. Both breeds showed enrichment for homophilic cell adhesion (GO:0007156), regulation of immune system process (GO:0002682), positive regulation of T cell activation (GO:0050870), and regulation of cytokine production (GO:0001817), highlighting immune-related and intercellular communication pathways. Royal White showed unique enrichment for immunoglobulin production (GO:0002381) and peptide antigen assembly (GO:0002503), suggesting possible stronger signals in adaptive immunity. In contrast, WD showed unique enrichment for neurodevelopmental and physiological processes, including axon guidance (GO:0007411), membrane depolarization during action potential (GO:0086010), and tissue development (GO:0009888), along with complement activation (GO:0006958), the latter indicating a potential innate immune component. Overall, functional enrichment analyses revealed shared impacts of HIGH and MODERATE impact variants in RW and WD sheep on immune function, cytoskeletal organization, and membrane-associated transport, alongside breed-specific putative signatures highlighting metabolic, immune-regulatory, and neurophysiological differences.
3.3. Population Structure Analysis
Population structure was assessed using both PCA and DAPC to explore genetic differentiation between breeds. As shown in Figure 5, the PCA plot (Panel A) displays the first two principal components, which explained 14.83% and 13.69% of the total genetic variation, respectively. While overlap between individuals from the two breeds is observed, breed-level clustering is evident, with RW and WD forming partially distinct groups along PC1. The 95% confidence ellipses further illustrate the separation pattern between breeds. To complement PCA, DAPC was performed to maximize between-breed variation. The density plot of the first discriminant function (Panel B) reveals a clearer distinction between RW and WD individuals, indicating that DAPC was able to enhance the separation observed in PCA. These results collectively suggest detectable but moderate genetic structuring between the two breeds.
3.4. Selective Sweep Analysis
Selective sweep regions were identified by integrating FST, nucleotide diversity (), and Tajima’s D metrics. Genes within these regions were annotated, and existing SNPs overlapping with sheep QTL records were mapped to known trait associations. Both RW and WD exhibited putative signatures of selection related to parasite resistance, but the underlying genes differed between breeds. In RW, parasite resistance signals involved TGFB2, TOX2, and HERC6, while in WD, they were associated with LAMC1, COLGALT2, TRIM14, and EPHA5. In addition to parasite resistance, RW showed putative selection on behavioral genes (GRM5, MAGI2), metabolic disease susceptibility (ALDH5A1), and growth- and quality-related loci (JADE2, PARP8, NIN, NRXN1) linked to body size, meat composition, milk production, and fiber-related characteristics (Table 5, Table 6). White Dorper displayed additional putative selection on growth loci (PLXDC2, HYDIN), milk composition (TENM2, BUD23, SCN8A), reproduction (STPG3, DYNC2H1), and morphology (LCN8, NFKB1) (Table 6). Collectively, these patterns indicate that while both breeds show putative selection for economically important traits and parasite resistance, RW may have stronger signals related to adaptive immunity and wool traits, whereas WD shows a broader balance across growth, reproduction, and parasite resistance.
4. Discussion
This study aimed to characterize genomic variations, assess population structure, and identify putative signatures of selection in two sheep breeds, RW and WD, which have distinct origins but share similar meat production purposes. Leveraging WGS data, we comprehensively analyzed SNP and indel variants, their functional impacts, and genomic regions under putative selection. Our findings provide valuable insights into the genetic architecture differentiating these breeds, with particular attention to loci related to parasite resistance, growth, and reproductive traits that may contribute to subtle phenotypic differences despite their overall production similarity.
4.1. Genomic Variant Characteristics
In the current study, WGS of 11 RW and 9 WD sheep identified approximately 21.96 million and 18.64 million SNPs and 2.87 million and 2.40 million indels, respectively. The sequencing coverage depth in this dataset ranged from 5.01× to 10×. These variant counts fall within the expected range when compared to other sheep WGS efforts. For example, a multi-species study involving domestic and wild sheep populations, including 18 domestic sheep and multiple wild relatives, reported 125.98 million SNPs and 13.04 million indels in total. Per-breed SNP counts ranged from approximately 13 million in European mouflon (n = 3) to 53 million in Asiatic mouflon (n = 16), with indel counts ranging from about 3 million to 7 million per breed. These samples were sequenced at coverage depths between 12.2× and 36.9× [22]. Furthermore, the Ts/Tv ratios were calculated as 2.30 for RW and 2.16 for WD in the current study. These values are consistent with typical mammalian genomes, which often fall within the range of 2.0 to 2.5, and specifically align with ratios reported in other sheep population genomic studies [23,24]. The consistency of these ratios further supports the high quality and accuracy of our SNP calls. Differences in variant counts across studies may be influenced by several factors. One key factor is sequencing depth, which affects the sensitivity of variant detection. Another important factor is sample size, as larger cohorts are more likely to capture rare and population-specific variants. On a broader scale, a larger study of 297 Duolang sheep identified 43.97 million SNPs and 6.50 million indels at approximately 13.35× coverage [25]. This higher variant yield is expected, given the substantially larger sample size and deeper sequencing depth, which together increase the power to detect both common and rare variants. The comparison underscores how sequencing depth and cohort size can influence variant discovery, and supports the interpretation that the variant counts observed in RW and WD are appropriate for the study design and technical parameters. Importantly, these results confirm that the sequencing and variant calling pipeline captured sufficient polymorphic sites for downstream analyses, including population structure, functional annotation, and sweep detection. Such comprehensive variant catalogs are essential for understanding the genetic basis of trait differentiation and provide a foundation for breed-specific genomic selection strategies.
4.2. Functional Annotation and Enrichment
Functional annotation using SnpEff revealed that the vast majority of SNPs and indels were located in non-coding regions, such as introns and intergenic regions, consistent with findings in other complex genomes, including those of sheep. This pattern has been well-documented in livestock genomics, where over 95% of detected variants typically lie outside coding regions due to the large proportion of non-exonic DNA in mammalian genomes [26,27]. Although the majority of SNPs and indels were located in non-coding regions (e.g., introns and intergenic areas), a smaller but biologically relevant subset occurred in coding regions or at exon-intron boundaries. These included missense, frameshift, splice site, and stop gain/loss variants, all of which are predicted to affect protein structure or gene regulation and may contribute to phenotypic variation [28,29]. Variants with predicted functional consequences are especially important in livestock genomics because they often underlie key traits like body weight and health. For example, body weight has been associated with specific SNPs and QTL regions in Merino sheep [30], while milk production traits have been linked to high-impact variants in crossbred dairy sheep [31]. Similarly, a genome-wide association study (GWAS) in meat sheep revealed associations of production traits such as birth weight, weaning weight, scan weight, and fat and muscle depth, alongside health traits including footrot and mastitis, demonstrating the polygenic and multifaceted nature of livestock traits [32]. The identification of predicted HIGH and MODERATE impact variants in both RW and WD sheep highlights changes primarily affecting protein-coding regions through amino acid substitutions, premature stop codons, or splice site disruptions. These findings suggest that selective processes, whether natural or artificial, continue to shape breed-specific genomic landscapes. Similar observations have been made across livestock species, where selection often acts on coding or regulatory variants to promote adaptation and improve performance traits [28].
Functional enrichment analyses based on KEGG pathways and GO terms revealed both shared and breed-specific biological processes in RW and WD sheep, providing insights into the molecular mechanisms underlying immunity, metabolism, and other key physiological traits. Specifically, KEGG pathway analysis showed that in both RW and WD breeds, enrichment of ABC transporters (oas02010) likely reflects roles in substrate transport and immune function, consistent with studies showing ABC transporter involvement in antigen processing in cattle [33]. ECM–receptor interaction (oas04512) is central to tissue remodeling and cellular communication; it is notably enriched in the ovine mammary gland during lactation, where it regulates epithelial cell adhesion and remodeling [34]. The complement and coagulation cascades (oas04610) pathway is a central component of innate immunity and has been shown to mediate early defense responses in sheep against Haemonchus contortus [35]. Retinol metabolism (oas00830), uniquely enriched in RW, has been associated with parasite resistance in sheep. Specifically, it elevated retinol, related gene expression correlates with resistance to Echinococcus granulosus infection [36]. The WD-specific enrichment for Staphylococcus aureus infection (oas05150) may reflect genetic adaptations linked to immune defense against bacterial pathogens. Similar KEGG enrichment was reported in bovine mammary gland transcriptome analyses, where the Staphylococcus aureus infection pathway (bta05150) was significantly enriched among genes differentially expressed in cows with subclinical Staphylococcus aureus mastitis [37]. Complementary to the KEGG results, GO term analysis further revealed shared and unique functional categories in RW and WD sheep, providing an additional layer of insight into the biological significance of the identified variants.
At the molecular function level, both RW and WD showed strong and highly significant enrichment for ATP binding (GO:0005524), protein binding (GO:0005515), calcium ion binding (GO:0005509), and ABC-type transporter activity (GO:0140359). The term ATP binding (GO:0005524) has also been detected in genome-wide selection scans in Dorper and Hu sheep, implicating it in growth and metabolic regulation [38]. Both breeds’ enrichment for protein binding (GO:0005515) and ATP binding (GO:0005524) have been reported in Chinese indigenous sheep adapted to warm climates, where these terms are functionally linked to metabolic regulation and heat loss mechanisms [39]. Calcium ion binding (GO:0005509), another shared term, has been observed in goat muscle development, with Leizhou goat fetal muscle studies showing differentially expressed genes enriched for this function [40]. ABC-type transporter activity (GO:0140359) underscores roles in membrane transport and detoxification; ABC transporters are well-characterized in veterinary pharmacology and pathogen defense, such as in drug absorption and xenobiotic handling [41]. Royal White-specific enrichment included transmembrane signaling receptor activity (GO:0004888), which involves membrane-bound receptors that detect extracellular cues. In wild cervids, this GO term was significantly enriched in antler-related genomic regions, suggesting a role in regulating tissue growth and regeneration [42]. Another RW-unique term, carbohydrate binding (GO:0030246), has direct ovine evidence from structural studies of the secretory glycoprotein SPS-40, which demonstrated specific carbohydrate-binding properties and conformational switching upon binding chitin-like oligosaccharides [43]. In WD, cadherin binding (GO:0045296), a function crucial for cell–cell adhesion, was significantly enriched, and has been identified in epigenomic studies of tissue remodeling in other mammals, such as cattle rumen during weaning [44]. White Dorper also showed unique enrichment for endopeptidase inhibitor activity (GO:0004866), which may imply regulation of proteolysis, a function critical in tissue remodeling and inflammation across vertebrates. This term has been reported in marine male fish Cyprinodon variegatus, where its expression was significantly altered following environmental chemical exposure, suggesting its sensitivity to physiological and environmental stressors [45].
In the cellular component category, enrichment was observed for structural elements such as collagen trimer (GO:0005581), myosin complex (GO:0016459), plasma membrane (GO:0005886), and extracellular matrix (GO:0031012) in both RW and WD sheep. Similar GO terms have been reported in transcriptomic studies of goats and sheep, where structural components are commonly enriched in tissues under selective pressure, including muscle and skin [46,47]. The enrichment of these cellular structures reflects a shared influence on genes involved in tissue organization, cellular architecture, and interactions between cells and their environment. Functional studies in livestock have shown that collagen and myosin-related components are essential for muscle fiber formation, extracellular support, and mechanotransduction processes that contribute to animal growth and performance [48]. Royal White sheep exhibited unique enrichment for the catenin complex (GO:0016342), microtubule organizing center (GO:0005815), and cytoskeleton (GO:0005856). The catenin complex, which is integral to adherens junctions and cell–cell adhesion, plays a key role in tissue integrity and differentiation. In transgenic mice expressing ovine -catenin, increased hair follicle density was observed, underscoring its potential impact on sheep morphological traits [49]. White Dorper sheep exhibited unique enrichment for the microtubule (GO:0005874). This cellular component has been directly linked to reproductive tissue function in avian livestock. In Sichuan white geese, microtubule (GO:0005874) enrichment was identified in ovarian tissue through integrated DNA methylation and transcriptomic analyses, implicating microtubule-associated genes such as EML6 in follicle growth and development [50].
Biological process terms highlighted both common and breed-specific functional enrichments in RW and WD sheep. Among the shared terms, homophilic cell adhesion (GO:0007156) was enriched in both breeds and has been reported among the top biological process GO terms in protoscoleces from sheep liver cystic echinococcosis cysts, accounting for 18% of annotated genes and involving plasma membrane adhesion molecules critical for cell–cell recognition during host–parasite interactions [51]. The enrichment of regulation of cytokine production (GO:0001817) and regulation of immune system process (GO:0002682) in both breeds is consistent with an ovine PBMC transcriptome study, where GO:0001817 was enriched post-vaccination and GO:0002682 was enriched post-adjuvant treatment, underscoring their central roles in immune activation [52]. Royal White-specific enrichment for immunoglobulin production (GO:0002381) and peptide antigen assembly (GO:0002503) points to emphasis on adaptive immunity. This is supported by deep sequencing of ovine immunoglobulin repertoires, which revealed extensive CDR3 diversity and active somatic hypermutation, indicating the capacity for enhanced and efficient humoral immune responses against infections [53]. In WD, unique enrichment was observed for primarily neurodevelopmental and excitability pathways, including axon guidance (GO:0007411) [54] and membrane depolarization during action potential (GO:0086010), processes also identified in livestock selection studies such as runs of homozygosity-based analyses in indigenous rabbit breeds [55]. While these terms are not classically immune-related, WD also exhibited unique enrichment for complement activation (GO:0006958), a core innate immune process. Complement activation has been reported as a key component of resistance to Haemonchus contortus infection in parasite-resistant sheep breeds (e.g., Canaria Hair Breed), linking this WD-specific term to protective immune functions [56].
4.3. Candidate Genes Under Selection
Selective sweep regions were identified by integrating population differentiation (FST), nucleotide diversity (), and Tajima’s D metrics, a commonly used integrative approach for detecting recent positive selection [57,58,59].
4.3.1. Candidate Genes in Royal White Sheep
In RW sheep, selective sweep analysis revealed putative candidate genes associated with a range of traits, including health, behavior, growth, meat quality, and milk production. Many of these genes overlapped with known QTLs and have functional support from studies in sheep or other species, suggesting potential roles in breed-specific adaptation and productivity.
Health traits: Genes in this category include NRXN1, HERC6, TGFB2, TOX2, and ALDH5A1. NRXN1 was identified in two differentiated regions, associated respectively with red blood cell distribution width (associated SNP: rs409057468) [60] and fiber diameter coefficient of variation (associated SNP: rs429232758) [61]. These traits are consistent with findings from QTL studies in Alpine Merino and fine-wool sheep breeds, suggesting pleiotropic effects on health and fleece quality. HERC6, linked to fecal egg count, has been implicated in the host response to parasitic infection in an Australian sheep population [62]. In addition, HERC6 has been associated with milk production, growth, and feed efficiency in various livestock populations [63,64]. TGFB2, located in a region containing two different SNPs (rs162057314 and rs160759291) on chromosome 12, was associated with resistance to gastrointestinal nematodes (Haemonchus contortus), supported by multiple studies of resistance loci in sheep and goats [65,66]. The SNP rs423531735, associated with immune regulation [67], maps to the gene TOX2. While its specific function in sheep immunity has yet to be explored, studies in mice and humans indicate that TOX2 is integral to germinal center T follicular helper (GC TFH) cell formation and memory responses [68], suggesting a cross-species role in adaptive immune function. The gene ALDH5A1 harbored a SNP (rs421181203) that was identified in sheep as significantly associated with susceptibility to Mycobacterium avium subsp. paratuberculosis and antibody titer levels, suggesting a potential role in immune defense [69]. Supporting evidence from dairy cattle indicates that ALDH5A1 expression is linked to antibody-mediated immune responses, as individuals with higher expression showed traits consistent with enhanced immunity [70]. Together, these findings suggest that ALDH5A1 may play a conserved role in pathogen resistance across ruminant species.
Behavior traits: Genes in this category include MAGI2 and GRM5. Notably, two SNPs (rs424244818 and rs424837012) located within the GRM5 gene and one SNP (rs429561404) within the MAGI2 gene overlapped with QTL associated with vocalization and locomotion responses, as identified in studies of social and handling reactivity in sheep [71]. Although experimental evidence for these two genes in sheep is limited, this positional overlap highlights their candidacy as behavior-related loci. In particular, GRM5, which encodes a glutamate metabotropic receptor, has been associated with movement patterns and grazing behavior in beef cattle [72,73], suggesting a conserved role in behavioral regulation across ruminants. For MAGI2, while functional studies in sheep are lacking, its reported association with feed efficiency in cattle implies potential relevance to broader physiological or behavioral traits [74].
Milk production trait: The gene NIN (Ninein) harbored SNP rs410734119, which overlapped a QTL associated with milk yield in sheep. Although this positional evidence suggests potential involvement in lactation traits, current functional studies across species have not linked NIN to milk production or mammary gland biology. NIN encodes a centrosomal protein involved in microtubule anchoring and epithelial cell organization, with well-characterized roles in neural development and cytoskeletal dynamics in humans and mice [75]. However, no direct evidence currently supports its role in lactation, either through gene expression profiling or functional assays. Further studies are needed to determine whether the observed association reflects a causal relationship, a regulatory linkage to nearby lactation-relevant genes, or an indirect positional effect.
Growth trait: The gene JADE2 overlapped a QTL linked to 6-month body weight, supported by genome-wide association studies in Baluchi sheep [76]. In Djallonké sheep, JADE2 was located within a copy number variation region (CNVR) hotspot associated with lipid metabolism traits, further suggesting its potential role in growth and energy regulation [77].
Meat trait: SNP rs416975775, which influences the omega-6 to omega-3 fatty acid ratio in sheep meat, is located within the gene PARP8 [78]. Although the direct involvement of PARP8 in meat traits in sheep remains unconfirmed, members of the same poly(ADP-ribose) polymerase gene family, such as PARP1, have been implicated in post-mortem muscle tenderization mechanisms [79], supporting a potential role for PARP8 in muscle-related phenotypes.
4.3.2. Candidate Genes in White Dorper Sheep
In WD sheep, selective sweep regions uncovered functionally relevant genes linked to growth, immunity, reproduction, and milk production traits.
Health traits: The genes TRIM14, COLGALT2, LAMC1, and EPHA5 were identified in sweep regions linked to immune-related traits. The gene TRIM14, containing SNP rs422296454, was identified within a selective sweep region in WD sheep and is associated with increased hematocrit levels during gastrointestinal nematode infection [80], which found the same SNP and the same gene in the current study. In humans, TRIM14 has been described as a regulator of innate immune signaling and a putative tumor suppressor, modulating interferon pathways in non-small cell lung cancer [81]. These findings indicate that TRIM14 is under selective pressure in sheep due to its immunological and potentially pleiotropic functions. The SNP rs402132699, located in COLGALT2, was identified in a Brazilian Morada Nova sheep study as being associated with hematocrit (packed cell volume after nematode challenge) and fecal egg count (fecal egg count after nematode challenge) [82]. Additionally, COLGALT2 was among several glycosyltransferases identified via GWAS as candidate loci for milk oligosaccharide synthesis in Holstein and Jersey cattle [83], suggesting its potential influence on the nutritional quality and functional properties of milk. Moreover, COLGALT2 has been shown to be overexpressed in human ovarian cancer, where it interacts with PLOD3, suggesting a role in collagen glycosylation and extracellular matrix organization [84]. Taken together, these cross-species findings indicate that selective pressure on COLGALT2 in sheep may reflect its conserved function in glycan metabolism, growth regulation, and tissue adaptation. The SNP rs430289425 (located in LAMC1) was identified as associated with resistance to Haemonchus contortus infection in sheep and goats [65]. In dairy cattle, a novel QTL was discovered near the LAMC1/2 locus (BTA16:63823597), which was associated with variation in teat width, suggesting a potential role in tissue organization and mammary gland morphology [85]. Similarly, SNP rs426828157 (located in the gene EPHA5) was identified as associated with low fecal egg count in sheep [86], indicating a potential role in parasite resistance. Although direct functional validation in immunity is limited from this work, previous studies have highlighted EPHA5 as a candidate gene for wool traits in Chinese Merino and Kirghiz sheep populations [87,88]. Additionally, in goats, EPHA5 has been associated with body length and implicated in insulin-mediated growth pathways [89], suggesting broader physiological functions across livestock species.
Milk production traits: Genes under selection included TENM2, BUD23, and SCN8A. SNP rs409487914 in TENM2 overlapped a QTL for milk fat yield in sheep. SNP rs430795622, associated with 180-day milk fat yield [31], was located in BUD23, a gene identified in the current study. Although no livestock-specific studies have directly linked BUD23 to milk traits, it encodes an 18S rRNA methyltransferase known to regulate mitochondrial function and lipid metabolism in mice and humans [90,91]. Given the high energy demands of milk synthesis, these roles suggest a plausible functional relevance of BUD23 to lactation performance in ruminants. The gene SCN8A, harboring SNP rs419496265, intersects with selective sweep signals and QTLs for milk fat percentage. Although its role in sheep lactation remains unconfirmed, livestock studies have demonstrated that SCN8A is expressed in spermatozoa, specifically localized to the flagellum and neck of mammalian sperm cells, and is associated with sperm motility traits in pigs and horses [92,93]. Given its involvement in cellular excitability and ion transport, SCN8A may contribute to broader physiological processes relevant to energy metabolism and secretory activity in ruminant tissues. SNP rs409487914 (located in TENM2) was associated with milk fat yield in sheep, although its specific role in ovine lactation remains unverified in any species, indicating a need for further research.
Growth traits: Selective sweep regions identified PLXDC2 and HYDIN as candidates linked to body weight. The SNP rs410323459, located in the gene HYDIN, was associated with 8-month body weight in Iranian sheep populations, suggesting its role in late-stage growth performance [94]. Similarly, SNP rs401963094, located in PLXDC2, was associated with body weight at 9 months in Lori-Bakhtiari sheep [95]. Additionally, PLXDC2 has been linked to reproductive traits in Holstein cattle [96], highlighting its broader developmental importance.
Meat traits: The gene ADD2, harboring SNP rs417859328, was located within a selective sweep region in WD sheep and was associated with dressing percentage [97]. While direct evidence linking ADD2 to meat traits in sheep is limited, its paralog ADD1 offers valuable insights. In beef cattle, multiple SNPs within ADD1 were significantly associated with growth traits and are potentially useful for marker-assisted selection in breeding programs [98]. Similarly, in pigs, polymorphisms in ADD1 were linked to meat quality differences between Meishan and other commercial breeds, suggesting that the adducin gene family plays a role in adipose and muscle development [99]. These findings support the hypothesis that adducins, including ADD2, may influence carcass-related traits in livestock through cytoskeletal regulation and tissue organization pathways.
Reproductive traits: Genes were represented by loci such as DYNC2H1 and STPG3. SNP rs413723884 (located in DYNC2H1) was identified in association with offspring number across four parities [100]. While direct functional studies of DYNC2H1 in livestock are unavailable, the gene is known in humans and mice to encode a cytoplasmic dynein heavy chain essential for retrograde intraflagellar transport in primary cilia, which is critical for Hedgehog and Wnt signaling pathways that regulate ovarian follicle development and reproductive tissue morphogenesis [101,102]. These conserved cellular functions support its potential involvement in sheep prolificacy, consistent with its positional mapping in reproductive trait QTL. The gene STPG3, harboring SNP rs430682724, was situated within a selective sweep region linked to litter size in sheep, overlapping QTL evidence for offspring number in global breeds [103], suggesting a role in prolificacy. It is also known to be abundantly expressed in the testes of both humans and mice, as identified in a CRISPR-based screening study targeting testis-enriched genes for contraceptive development [104]. Although knockout of STPG3 did not impair male fertility in mice, its high expression in reproductive tissues supports a potential role in gametogenesis or sperm function. Additionally, a related gene, STPG2, has been implicated in male infertility, specifically azoospermia, in a Taiwanese cohort study investigating microtubule-associated gene clusters [105]. These findings suggest that STPG3, while not essential for male fecundity in mice, may participate in conserved testis-specific pathways that are potentially relevant to sheep reproduction.
Morphological traits: Genes under selection included LCN8 and NFKB1. The LCN8 gene, identified in a selective sweep region and associated with horn number through SNP rs415039972 [103], presents an interesting case of potential pleiotropy or positional linkage. Although its QTL association relates to horn phenotype, functional studies primarily describe LCN8 as a member of the lipocalin family involved in male reproduction. In sheep, LCN8 is highly expressed in the caput epididymis, where spermatozoa begin maturation [106]. Similarly, in humans and other mammals, it is enriched in the corpus region of the epididymis, suggesting a conserved role in sperm development and epididymal function [107]. This apparent functional divergence may indicate a pleiotropic influence or a neighboring regulatory element that affects both horn development and reproductive traits, which warrants further functional validation. Morphological selection was further supported by NFKB1, identified in a selective sweep region containing SNP rs416625889, which has been associated with bone area in QTL mapping studies of Scottish Blackface lambs [108]. Although primarily known for its role in immune regulation, NFKB1 is a key transcription factor in the nuclear factor kappa B signaling pathway, which mediates inflammatory responses and cellular stress signaling. In sheep, NFKB1 is actively expressed in maternal inguinal lymph nodes during early pregnancy, indicating its involvement in immune modulation at the maternal–fetal interface [109]. Furthermore, a retrospective SNP analysis of host resistance and susceptibility to ovine Johne’s disease, caused by Mycobacterium avium subsp. paratuberculosis, identified significant variants near genes involved in immune-related pathways, including the nuclear factor kappa B and mitogen-activated protein kinase signaling pathways, underscoring their role in host defense mechanisms against infection [110]. Broader livestock studies also implicate NFKB1 as a key transcription factor in the regulation of immune and inflammatory responses, playing a major role in mastitis susceptibility in beef cattle [111]. These findings suggest that NFKB1 may influence multiple traits under selection, including immune function, growth, and tissue development, either directly or through pleiotropic effects.
4.4. Limitations and Future Directions
This study provides foundational information on the genomic architecture of RW and WD sheep; however, several limitations should be acknowledged. The relatively small sample size may have limited the detection of rare variants and reduced the power to identify subtle signals of selection. Candidate genes were prioritized based on the presence or overlap of SNPs, recorded in the Sheep QTL Database, that were located within identified selective sweep regions; however, functional validation of these genes is still lacking. Future research should include larger and more diverse populations across multiple breeds, integrate gene expression and functional assays, and broaden variant discovery to encompass structural variants and epigenetic modifications, thereby enabling a more comprehensive understanding of breed-specific adaptations.
5. Conclusions
In conclusion, this study presents the first WGS-based comparative analysis between RW and WD sheep, identifying over 40 million SNPs and 5 million indels across the two breeds. Through variant annotation, population structure analyses, and selective sweep detection, we revealed breed-specific genomic regions and candidate genes associated with traits such as health, reproduction, growth, and production. These findings enhance our understanding of genetic differentiation between hair sheep breeds and provide a valuable foundation for future research and genomic selection in sheep breeding programs. Furthermore, the variants identified in this work can be investigated in additional populations and incorporated into genome-wide association studies to further elucidate the genetic architecture of economically important traits, ultimately supporting selective breeding strategies aimed at improving production and health in these breeds.
Author Contributions
Conceptualization, M.L., A.K., D.H., N.S., and R.C.; methodology, M.L., A.K., D.H., N.S., and R.C.; software, M.L. and D.H.; validation, M.L.; resources, A.K. and N.S.; data curation, A.K. and N.S.; writing—original draft preparation, M.L.; writing—review and editing, M.L., A.K., N.S., D.H., and R.C.; visualization, M.L.; supervision, R.C. and N.S.; project administration, R.C. and N.S.; funding acquisition, N.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Virginia Maryland College of Veterinary Medicine Internal Research Funding through the Department of Biomedical Sciences and Pathobiology.
Institutional Review Board Statement
All animal samples were approved by the Virginia Polytechnic Institute and State University Institutional Animal Care and Use Committee (IACUC) #17-233 and Institutional Biosafety Committee (IBC) # 20-067.
Informed Consent Statement
Not applicable
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to legal restrictions.
Acknowledgments
We would like to thank Dr. Michael Collins of the University of Wisconsin–Madison School of Veterinary Medicine for his invaluable contribution to the identification of the flock and the facilitation of collaboration to obtain the samples.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Food and Agriculture Organization of the United Nations. FAOSTAT: Live animals – sheep. https://www.fao.org/faostat/en/#data/QA, 2024. Accessed January 31, 2026.
- United States Department of Agriculture, National Agricultural Statistics Service. Sheep and Goats. https://downloads.usda.library.cornell.edu/usda-esmis/files/000000018/zk51xc07n/9593wq66x/shep0125.pdf?utm_source=chatgpt.com, 2025. Released January 2025, Accessed June 19, 2025.
- Royal White® Sheep. Royal White® Sheep – Official Breed Information. https://royalwhitesheep.biz/, 2024. Accessed June 19, 2025.
- Milne, C. The history of the Dorper sheep. Small Ruminant Research 2000, 36, 99–102. [CrossRef]
- Ojango, J.M.; Okpeku, M.; Osei-Amponsah, R.; Kugonza, D.R.; Mwai, O.; Changunda, M.G.; Olori, V.E. Dorper sheep in Africa: a review of their use and performance in different environments. CABI Reviews 2023.
- Wanjala, G.; Astuti, P.K.; Bagi, Z.; Kichamu, N.; Strausz, P.; Kusza, S. Assessing the genomics structure of Dorper and white Dorper variants, and Dorper populations in South Africa and Hungary. Biology 2023, 12, 386. [CrossRef]
- Gavojdian, D.; Budai, C.; Cziszter, L.T.; Csizmar, N.; Jávor, A.; Kusza, S. Reproduction efficiency and health traits in Dorper, White Dorper, and Tsigai sheep breeds under temperate European conditions. Asian-Australasian journal of animal sciences 2015, 28, 599. [CrossRef]
- Daetwyler, H.D.; Capitan, A.; Pausch, H.; Stothard, P.; Van Binsbergen, R.; Brøndum, R.F.; Liao, X.; Djari, A.; Rodriguez, S.C.; Grohs, C.; et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nature genetics 2014, 46, 858–865. [CrossRef]
- Hayes, B.J.; Daetwyler, H.D.; Bowman, P.J.; Chamberlian, A.; Vander Jagt, C.; Capitan, A.; Pausch, H.; Stothard, P.; Liao, X.; Schrooten, C.; et al. Genomic prediction from whole genome sequence in livestock: the 1000 bull genomes project. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production. American Society of Animal Science, 2014.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 2013. [CrossRef]
- Van der Auwera, G.A.; O’Connor, B.D. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Current protocols in bioinformatics 2020, 43, 10–11. [CrossRef]
- Li, H. Statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [CrossRef]
- Chen, H.; Boutros, P. VennDiagram: Generate High-Resolution Venn and Euler Plots, 2011. R package version 1.7.3.
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 2009, 4, 44–57. [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics 2018, 210, 767–777. [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 2007, 81, 559–575. [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria, 2023.
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics 2010, 11, 94. [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [CrossRef]
- Chen, Z.H.; Xu, Y.X.; Xie, X.L.; Wang, D.F.; Aguilar-Gómez, D.; Liu, G.J.; Li, X.; Esmailizadeh, A.; Rezaei, V.; Kantanen, J.; et al. Whole-genome sequence analysis unveils different origins of European and Asiatic mouflon and domestication-related genes in sheep. Communications Biology 2021, 4, 1307. [CrossRef]
- Tian, D.; Han, B.; Li, X.; Liu, D.; Zhou, B.; Zhao, C.; Zhang, N.; Wang, L.; Pei, Q.; Zhao, K. Genetic diversity and selection of Tibetan sheep breeds revealed by whole-genome resequencing. Animal bioscience 2023, 36, 991. [CrossRef]
- Yao, Y.; Pan, Z.; Di, R.; Liu, Q.; Hu, W.; Guo, X.; He, X.; Gan, S.; Wang, X.; Chu, M. Whole genome sequencing reveals the effects of recent artificial selection on litter size of Bamei mutton sheep. Animals 2021, 11, 157. [CrossRef]
- Fang, C.; Druet, T.; Cao, H.; Liu, W.; Chen, Q.; Farnir, F. Whole genome sequences of 297 Duolang sheep for litter size. Scientific Data 2025, 12, 1086. [CrossRef]
- Ma, R.; Kuang, R.; Zhang, J.; Sun, J.; Xu, Y.; Zhou, X.; Han, Z.; Hu, M.; Wang, D.; Fu, Y.; et al. Annotation and assessment of functional variants in livestock through epigenomic data. Journal of Genetics and Genomics 2025. [CrossRef]
- Wang, Z.H.; Zhu, Q.H.; Li, X.; Zhu, J.W.; Tian, D.M.; Zhang, S.S.; Kang, H.L.; Li, C.P.; Dong, L.L.; Zhao, W.M.; et al. iSheep: an integrated resource for sheep genome, variant and phenotype. Frontiers in Genetics 2021, 12, 714852. [CrossRef]
- Guo, Y.; Liang, J.; Lv, C.; Wang, Y.; Wu, G.; Ding, X.; Quan, G. Sequencing reveals population structure and selection signatures for reproductive traits in Yunnan semi-fine wool sheep (Ovis aries). Frontiers in Genetics 2022, 13, 812753. [CrossRef]
- Kijas, J.W.; Lenstra, J.A.; Hayes, B.; Boitard, S.; Porto Neto, L.R.; San Cristobal, M.; Servin, B.; McCulloch, R.; Whan, V.; Gietzen, K.; et al. Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS biology 2012, 10, e1001258. [CrossRef]
- Al-Mamun, H.A.; Kwan, P.; Clark, S.A.; Ferdosi, M.H.; Tellam, R.; Gondro, C. Genome-wide association study of body weight in Australian Merino sheep reveals an orthologous region on OAR6 to human and bovine genomic regions affecting height and weight. Genetics Selection Evolution 2015, 47, 66. [CrossRef]
- Li, H.; Wu, X.L.; Tait Jr, R.; Bauck, S.; Thomas, D.; Murphy, T.; Rosa, G. Genome-wide association study of milk production traits in a crossbred dairy sheep population using three statistical models. Animal Genetics 2020, 51, 624–628. [CrossRef]
- Kaseja, K.; Mucha, S.; Yates, J.; Smith, E.; Banos, G.; Conington, J. Genome-wide association study of health and production traits in meat sheep. animal 2023, 17, 100968. [CrossRef]
- Lopez, B.I.; Santiago, K.G.; Lee, D.; Ha, S.; Seo, K. RNA sequencing (RNA-Seq) based transcriptome analysis in immune response of Holstein cattle to killed vaccine against bovine viral diarrhea virus type I. Animals 2020, 10, 344. [CrossRef]
- Chen, W.; Gu, X.; Lv, X.; Cao, X.; Yuan, Z.; Wang, S.; Sun, W. Non-coding transcriptomic profiles in the sheep mammary gland during different lactation periods. Frontiers in veterinary science 2022, 9, 983562. [CrossRef]
- Lins, J.G.G.; Amarante, A.F. Complement and Coagulation Cascade Activation Regulates the Early Inflammatory Mechanism of Resistance of Suckling Lambs Against Haemonchus contortus. Pathogens 2025, 14, 447. [CrossRef]
- Li, X.; Jiang, S.; Wang, X.; Jia, B. Intestinal transcriptomes in Kazakh sheep with different haplotypes after experimental Echinococcus granulosus infection. Parasite 2021, 28, 14. [CrossRef]
- Wang, M.; Bissonnette, N.; Laterrière, M.; Dudemaine, P.L.; Gagné, D.; Roy, J.P.; Sirard, M.A.; Ibeagha-Awemu, E.M. Gene co-expression in response to Staphylococcus aureus infection reveals networks of genes with specific functions during bovine subclinical mastitis. Journal of Dairy Science 2023, 106, 5517–5536. [CrossRef]
- Lv, X.; Chen, W.; Wang, S.; Cao, X.; Yuan, Z.; Getachew, T.; Mwacharo, J.M.; Haile, A.; Sun, W. Whole-genome resequencing of Dorper and Hu sheep to reveal selection signatures associated with important traits. Animal Biotechnology 2023, 34, 3016–3026. [CrossRef]
- Jin, M.; Wang, H.; Liu, G.; Lu, J.; Yuan, Z.; Li, T.; Liu, E.; Lu, Z.; Du, L.; Wei, C. Whole-genome resequencing of Chinese indigenous sheep provides insight into the genetic basis underlying climate adaptation. Genetics Selection Evolution 2024, 56, 26. [CrossRef]
- Ye, J.; Zhao, X.; Xue, H.; Zou, X.; Liu, G.; Deng, M.; Sun, B.; Guo, Y.; Liu, D.; Li, Y. RNA-Seq reveals miRNA and mRNA co-regulate muscle differentiation in fetal Leizhou goats. Frontiers in Veterinary Science 2022, 9, 829769. [CrossRef]
- How, S.S.; Nathan, S.; Lam, S.D.; Chieng, S. ATP-binding cassette (ABC) transporters: structures and roles in bacterial pathogenesis. Journal of Zhejiang University-Science B 2025, 26, 58–75. [CrossRef]
- Anderson, S.; Côté, S.; Richard, J.; Shafer, A. Genomic architecture of phenotypic extremes in a wild cervid. BMC genomics 2022, 23, 126. [CrossRef]
- Srivastava, D.B.; Ethayathulla, A.S.; Kumar, J.; Somvanshi, R.K.; Sharma, S.; Dey, S.; Singh, T.P. Carbohydrate binding properties and carbohydrate induced conformational switch in sheep secretory glycoprotein (SPS-40): Crystal structures of four complexes of SPS-40 with chitin-like oligosaccharides. Journal of structural biology 2007, 158, 255–266. [CrossRef]
- Boschiero, C.; Gao, Y.; Baldwin, R.L.; Ma, L.; Liu, G.E.; Li, C.J. Characterization of accessible chromatin regions in cattle rumen epithelial tissue during weaning. Genes 2022, 13, 535. [CrossRef]
- Schönemann, A.M.; Beiras, R.; Diz, A.P. Widespread alterations upon exposure to the estrogenic endocrine disruptor ethinyl estradiol in the liver proteome of the marine male fish Cyprinodon variegatus. Aquatic Toxicology 2022, 248, 106189. [CrossRef]
- Hou, X.; Wang, X.; Hou, S.; Dang, J.; Zhang, X.; Tang, J.; Shi, Y.; Ma, S.; Xu, Z. Comparative ultrastructural and transcriptomic profile analysis of skin tissues from indigenous, improved meat, and dairy goat breeds. BMC genomics 2024, 25, 1070. [CrossRef]
- Li, J.; Chen, C.; Zhao, R.; Wu, J.; Li, Z. Transcriptome analysis of mRNAs, lncRNAs, and miRNAs in the skeletal muscle of Tibetan chickens at different developmental stages. Frontiers in Physiology 2023, 14, 1225349. [CrossRef]
- Tsang, H.G.; Clark, E.L.; Markby, G.R.; Bush, S.J.; Hume, D.A.; Corcoran, B.M.; MacRae, V.E.; Summers, K.M. Expression of calcification and extracellular matrix genes in the cardiovascular system of the healthy domestic sheep (Ovis aries). Frontiers in Genetics 2020, 11, 919. [CrossRef]
- Wang, J.; Cui, K.; Hua, G.; Han, D.; Yang, Z.; Li, T.; Yang, X.; Zhang, Y.; Cai, G.; Deng, X.; et al. Skin-specific transgenic overexpression of ovine β-catenin in mice. Frontiers in Genetics 2023, 13, 1059913. [CrossRef]
- Ma, L.; Zhao, X.; Wang, H.; Chen, Z.; Zhang, K.; Xue, J.; Luo, Y.; Liu, H.; Jiang, X.; Wang, J.; et al. DNA Methylation Patterns and Transcriptomic Data Were Integrated to Investigate Candidate Genes Influencing Reproductive Traits in Ovarian Tissue from Sichuan White Geese. International Journal of Molecular Sciences 2025, 26, 3408. [CrossRef]
- Pereira, I.; Hidalgo, C.; Stoore, C.; Baquedano, M.S.; Cabezas, C.; Bastías, M.; Riveros, A.; Meneses, C.; Cancela, M.; Ferreira, H.B.; et al. Transcriptome analysis of Echinococcus granulosus sensu stricto protoscoleces reveals differences in immune modulation gene expression between cysts found in cattle and sheep. Veterinary Research 2022, 53, 8. [CrossRef]
- Varela-Martínez, E.; Abendaño, N.; Asín, J.; Sistiaga-Poveda, M.; Pérez, M.M.; Reina, R.; De Andres, D.; Luján, L.; Jugo, B.M. Molecular signature of aluminum hydroxide adjuvant in ovine PBMCs by integrated mRNA and microRNA transcriptome sequencing. Frontiers in immunology 2018, 9, 2406. [CrossRef]
- Park, M.; de Villavicencio Diaz, T.N.; Lange, V.; Wu, L.; Le Bihan, T.; Ma, B. Exploring the sheep (Ovis aries) immunoglobulin repertoire by next generation sequencing. Molecular Immunology 2023, 156, 20–30. [CrossRef]
- Kumar, P.; Becker, J.C.; Gao, K.; Carney, R.P.; Lankford, L.; Keller, B.A.; Herout, K.; Lam, K.S.; Farmer, D.L.; Wang, A. Neuroprotective effect of placenta-derived mesenchymal stromal cells: role of exosomes. The FASEB Journal 2019, 33, 5836. [CrossRef]
- Ping, X.; Chen, Y.; Wang, H.; Jin, Z.; Duan, Q.; Ren, Z.; Dong, X. Whole-genome sequencing reveals patterns of runs of homozygosity underlying genetic diversity and selection in domestic rabbits. BMC genomics 2025, 26, 425. [CrossRef]
- Guo, Z.; González, J.F.; Hernandez, J.N.; McNeilly, T.N.; Corripio-Miyar, Y.; Frew, D.; Morrison, T.; Yu, P.; Li, R.W. Possible mechanisms of host resistance to Haemonchus contortus infection in sheep breeds native to the Canary Islands. Scientific reports 2016, 6, 26200. [CrossRef]
- Yang, J.; Li, W.R.; Lv, F.H.; He, S.G.; Tian, S.L.; Peng, W.F.; Sun, Y.W.; Zhao, Y.X.; Tu, X.L.; Zhang, M.; et al. Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Molecular biology and evolution 2016, 33, 2576–2592. [CrossRef]
- Sun, L.; Yuan, C.; Guo, T.; Zhang, M.; Bai, Y.; Lu, Z.; Liu, J. Resequencing reveals population structure and genetic diversity in Tibetan sheep. BMC genomics 2024, 25, 906. [CrossRef]
- Mei, C.; Gui, L.; Hong, J.; Raza, S.H.A.; Aorigele, C.; Tian, W.; Garcia, M.; Xin, Y.; Yang, W.; Zhang, S.; et al. Insights into adaption and growth evolution: a comparative genomics study on two distinct cattle breeds from Northern and Southern China. Molecular Therapy Nucleic Acids 2021, 23, 959–967. [CrossRef]
- Zhu, S.; Guo, T.; Zhao, H.; Qiao, G.; Han, M.; Liu, J.; Yuan, C.; Wang, T.; Li, F.; Yue, Y.; et al. Genome-wide association study using individual single-nucleotide polymorphisms and haplotypes for erythrocyte traits in Alpine Merino sheep. Frontiers in genetics 2020, 11, 848. [CrossRef]
- Zhao, H.; Guo, T.; Lu, Z.; Liu, J.; Zhu, S.; Qiao, G.; Han, M.; Yuan, C.; Wang, T.; Li, F.; et al. Genome-wide association studies detects candidate genes for wool traits by re-sequencing in Chinese fine-wool sheep. BMC genomics 2021, 22, 127. [CrossRef]
- Al Kalaldeh, M.; Gibson, J.; Lee, S.H.; Gondro, C.; Van Der Werf, J.H. Detection of genomic regions underlying resistance to gastrointestinal parasites in Australian sheep. Genetics Selection Evolution 2019, 51, 37. [CrossRef]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC genomics 2019, 20, 294. [CrossRef]
- Cheng, J.; Cao, X.; Hanif, Q.; Pi, L.; Hu, L.; Huang, Y.; Lan, X.; Lei, C.; Chen, H. Integrating genome-wide CNVs into QTLs and high confidence GWAScore regions identified positional candidates for sheep economic traits. Frontiers in Genetics 2020, 11, 569. [CrossRef]
- Estrada-Reyes, Z.M.; Tsukahara, Y.; Amadeu, R.R.; Goetsch, A.L.; Gipson, T.A.; Sahlu, T.; Puchala, R.; Wang, Z.; Hart, S.P.; Mateescu, R.G. Signatures of selection for resistance to Haemonchus contortus in sheep and goats. BMC genomics 2019, 20, 735. [CrossRef]
- Rodrigues, J.L.; Braga, L.G.; Watanabe, R.N.; Schenkel, F.S.; Berry, D.P.; Buzanskas, M.E.; Munari, D.P. Genetic diversity and selection signatures in sheep breeds. Journal of Applied Genetics 2025, pp. 1–13. [CrossRef]
- Sallé, G.; Jacquiet, P.; Gruner, L.; Cortet, J.; Sauvé, C.; Prévot, F.; Grisez, C.; Bergeaud, J.P.; Schibler, L.; Tircazes, A.; et al. A genome scan for QTL affecting resistance to Haemonchus contortus in sheep. Journal of animal science 2012, 90, 4690–4705. [CrossRef]
- Horiuchi, S.; Wu, H.; Liu, W.C.; Schmitt, N.; Provot, J.; Liu, Y.; Bentebibel, S.E.; Albrecht, R.A.; Schotsaert, M.; Forst, C.V.; et al. Tox2 is required for the maintenance of GC TFH cells and the generation of memory TFH cells. Science advances 2021, 7, eabj1249. [CrossRef]
- Usai, M.G.; Casu, S.; Sechi, T.; Salaris, S.L.; Miari, S.; Mulas, G.; Cancedda, M.G.; Ligios, C.; Carta, A. Advances in understanding the genetic architecture of antibody response to paratuberculosis in sheep by heritability estimate and LDLA mapping analyses and investigation of candidate regions using sequence-based data. Genetics Selection Evolution 2024, 56, 5. [CrossRef]
- Thompson-Crispi, K.A.; Sargolzaei, M.; Ventura, R.; Abo-Ismail, M.; Miglior, F.; Schenkel, F.; Mallard, B.A. A genome-wide association study of immune response traits in Canadian Holstein cattle. BMC genomics 2014, 15, 559. [CrossRef]
- Hazard, D.; Moreno, C.; Foulquié, D.; Delval, E.; François, D.; Bouix, J.; Sallé, G.; Boissy, A. Identification of QTLs for behavioral reactivity to social separation and humans in sheep using the OvineSNP50 BeadChip. BMC genomics 2014, 15, 778. [CrossRef]
- Moreno García, C.A.; Zhou, H.; Altimira, D.; Dynes, R.; Gregorini, P.; Jayathunga, S.; Maxwell, T.M.; Hickford, J. The glutamate metabotropic receptor 5 (GRM5) gene is associated with beef cattle home range and movement tortuosity. Journal of Animal Science and Biotechnology 2022, 13, 111. [CrossRef]
- Moreno García, C.A.; Perelman, S.B.; Dynes, R.; Maxwell, T.M.; Zhou, H.; Hickford, J. Key Grazing Behaviours of Beef Cattle Identify Specific Genotypes of the Glutamate Metabotropic Receptor 5 Gene (GRM5). Behavior Genetics 2024, 54, 212–229. [CrossRef]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasbarre, L.C.; Van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Functional & integrative genomics 2012, 12, 81–92.
- Moss, D.K.; Bellett, G.; Carter, J.M.; Liovic, M.; Keynton, J.; Prescott, A.R.; Lane, E.B.; Mogensen, M.M. Ninein is released from the centrosome and moves bi-directionally along microtubules. Journal of cell science 2007, 120, 3064–3074. [CrossRef]
- Gholizadeh, M.; Rahimi-Mianji, G.; Nejati-Javaremi, A. Genomewide association study of body weight traits in Baluchi sheep. Journal of Genetics 2015, 94, 143–146. [CrossRef]
- Goyache, F.; Fernández, I.; Tapsoba, A.S.R.; Traoré, A.; Menéndez-Arias, N.A.; Álvarez, I. Functional characterization of Copy Number Variations regions in Djallonké sheep. Journal of Animal Breeding and Genetics 2021, 138, 600–612. [CrossRef]
- Rovadoscki, G.; Pertile, S.; Alvarenga, A.; Cesar, A.; Pértille, F.; Petrini, J.; Franzo, V.; Soares, W.; Morota, G.; Spangler, M.L.; et al. Estimates of genomic heritability and genome-wide association study for fatty acids profile in Santa Inês sheep. BMC genomics 2018, 19, 375. [CrossRef]
- Li, R.; Luo, R.; Luo, Y.; Hou, Y.; Wang, J.; Zhang, Q.; Chen, X.; Hu, L.; Zhou, J. Biological function, mediate cell death pathway and their potential regulated mechanisms for post-mortem muscle tenderization of PARP1: A review. Frontiers in Nutrition 2022, 9, 1093939. [CrossRef]
- Thorne, J.W.; Redden, R.; Bowdridge, S.A.; Becker, G.M.; Stegemiller, M.R.; Murdoch, B.M. Genome-wide analysis of sheep artificially or naturally infected with gastrointestinal nematodes. Genes 2023, 14, 1342. [CrossRef]
- Hai, J.; Zhu, C.Q.; Wang, T.; Organ, S.L.; Shepherd, F.A.; Tsao, M.S. TRIM14 is a putative tumor suppressor and regulator of innate immune response in non-small cell lung cancer. Scientific reports 2017, 7, 39692. [CrossRef]
- Haehling, M.B.; Cruvinel, G.G.; Toscano, J.H.; Giraldelo, L.A.; Santos, I.B.; Esteves, S.N.; Benavides, M.V.; Júnior, W.B.; Niciura, S.C.; Chagas, A.C.S. Four single nucleotide polymorphisms (SNPs) are associated with resistance and resilience to Haemonchus contortus in Brazilian Morada Nova sheep. Veterinary Parasitology 2020, 279, 109053. [CrossRef]
- Poulsen, N.A.; Robinson, R.C.; Barile, D.; Larsen, L.B.; Buitenhuis, B. A genome-wide association study reveals specific transferases as candidate loci for bovine milk oligosaccharides synthesis. BMC genomics 2019, 20, 404. [CrossRef]
- Guo, T.; Li, B.; Kang, Y.; Gu, C.; Fang, F.; Chen, X.; Liu, X.; Lu, G.; Feng, C.; Xu, C. COLGALT2 is overexpressed in ovarian cancer and interacts with PLOD3. Clinical and Translational Medicine 2021, 11, e370. [CrossRef]
- Miles, A.M.; Posbergh, C.J.; Huson, H.J. Direct phenotyping and principal component analysis of type traits implicate novel QTL in bovine mastitis through genome-wide association. Animals 2021, 11, 1147. [CrossRef]
- Atlija, M.; Arranz, J.J.; Martinez-Valladares, M.; Gutiérrez-Gil, B. Detection and replication of QTL underlying resistance to gastrointestinal nematodes in adult sheep using the ovine 50K SNP array. Genetics Selection Evolution 2016, 48, 4. [CrossRef]
- Yang, R.; Han, Z.; Zhou, W.; Li, X.; Zhang, X.; Zhu, L.; Wang, J.; Li, X.; Zhang, C.l.; Han, Y.; et al. Population structure and selective signature of Kirghiz sheep by Illumina Ovine SNP50 BeadChip. PeerJ 2024, 12, e17980. [CrossRef]
- Wang, Z.; Zhang, H.; Yang, H.; Wang, S.; Rong, E.; Pei, W.; Li, H.; Wang, N. Genome-wide association study for wool production traits in a Chinese Merino sheep population. PloS one 2014, 9, e107101. [CrossRef]
- Moaeen-ud Din, M.; Danish Muner, R.; Khan, M.S. Genome wide association study identifies novel candidate genes for growth and body conformation traits in goats. Scientific Reports 2022, 12, 9891. [CrossRef]
- Martinez-Sanchez, N.; Brümmer, A.; Barron, N.J.; Rosoff, D.B.; Liechti, A.; Voronkov, M.; Hayter, E.A.; Chamois, S.; Dreos, R.; Guex, N.; et al. The 18S rRNA methyltransferase, BUD23, is required for appropriate lipid and mitochondrial metabolism. bioRxiv 2025, pp. 2025–05.
- Baxter, M.; Voronkov, M.; Poolman, T.; Galli, G.; Pinali, C.; Goosey, L.; Knight, A.; Krakowiak, K.; Maidstone, R.; Iqbal, M.; et al. Cardiac mitochondrial function depends on BUD23 mediated ribosome programming. Elife 2020, 9, e50705. [CrossRef]
- Gmel, A.I.; Burger, D.; Neuditschko, M. A novel QTL and a candidate gene are associated with the progressive motility of Franches-Montagnes stallion spermatozoa after thaw. Genes 2021, 12, 1501. [CrossRef]
- Marques, D.B.; Bastiaansen, J.W.; Broekhuijse, M.L.; Lopes, M.S.; Knol, E.F.; Harlizius, B.; Guimarães, S.E.; Silva, F.F.; Lopes, P.S. Weighted single-step GWAS and gene network analysis reveal new candidate genes for semen traits in pigs. Genetics Selection Evolution 2018, 50, 40. [CrossRef]
- Pasandideh, M.; Gholizadeh, M.; Rahimi-Mianji, G. A genome-wide association study revealed five SNPs affecting 8-month weight in sheep. Animal genetics 2020, 51, 973–976. [CrossRef]
- Almasi, M.; Zamani, P.; Mirhoseini, S.Z.; Moradi, M.H. Genome-wide association study for postweaning weight traits in Lori-Bakhtiari sheep. Tropical Animal Health and Production 2021, 53, 163. [CrossRef]
- Maddahi, N.; Sadeghi, M.; Miraee Ashtiani, S.R.; Kholghi, M.; Jalil Sarghale, A. Genome-wide association studies and candidate genes networks affecting reproductive traits using Iranian Holstein sequence data. BMC genomics 2025, 26, 656. [CrossRef]
- Ladeira, G.C.; Pilonetto, F.; Fernandes, A.C.; Bóscollo, P.P.; Dauria, B.D.; Titto, C.G.; Coutinho, L.L.; e Silva, F.F.; Pinto, L.F.B.; Mourão, G.B. CNV detection and their association with growth, efficiency and carcass traits in Santa Inês sheep. Journal of Animal Breeding and Genetics 2022, 139, 476–487. [CrossRef]
- Huang, Y.Z.; Qian, L.N.; Wang, J.; Zhang, C.L.; Fang, X.T.; Lei, C.Z.; Lan, X.Y.; Ma, Y.; Bai, Y.Y.; Lin, F.P.; et al. Genetic variants in ADD1 gene and their associations with growth traits in cattle. Animal Biotechnology 2019, 30, 7–12. [CrossRef]
- Li, C.; Pan, Y.; Me, H. Polymorphism of the H-FABP, MC4R and ADD1 genes in the Meishan and four other pig populations in China. South African Journal of Animal Science 2006, 36, 1–6. [CrossRef]
- Gholizadeh, M.; Rahimi-Mianji, G.; Nejati-Javaremi, A.; De Koning, D.J.; Jonas, E. Genomewide association study to detect QTL for twinning rate in Baluchi sheep. Journal of genetics 2014, 93, 489–493. [CrossRef]
- Ocbina, P.J.R.; Tuson, M.; Anderson, K.V. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PloS one 2009, 4, e6839. [CrossRef]
- Ocbina, P.J.R.; Eggenschwiler, J.T.; Moskowitz, I.; Anderson, K.V. Complex interactions between genes controlling trafficking in primary cilia. Nature genetics 2011, 43, 547–553. [CrossRef]
- Salehian-Dehkordi, H.; Xu, Y.X.; Xu, S.S.; Li, X.; Luo, L.Y.; Liu, Y.J.; Wang, D.F.; Cao, Y.H.; Shen, M.; Gao, L.; et al. Genome-wide detection of copy number variations and their association with distinct phenotypes in the world’s sheep. Frontiers in genetics 2021, 12, 670582. [CrossRef]
- Park, S.; Shimada, K.; Fujihara, Y.; Xu, Z.; Shimada, K.; Larasati, T.; Pratiwi, P.; Matzuk, R.M.; Devlin, D.J.; Yu, Z.; et al. CRISPR/Cas9-mediated genome-edited mice reveal 10 testis-enriched genes are dispensable for male fecundity. Biology of Reproduction 2020, 103, 195–204. [CrossRef]
- Chan, C.C.; Yen, T.H.; Tseng, H.C.; Mai, B.; Ho, P.K.; Chou, J.L.; Wu, G.J.; Huang, Y.C. A comprehensive genetic study of microtubule-associated gene clusters for male infertility in a Taiwanese cohort. International Journal of Molecular Sciences 2023, 24, 15363. [CrossRef]
- Wu, C.; Wang, C.; Zhai, B.; Zhao, Y.; Zhao, Z.; Yuan, Z.; Fu, X.; Zhang, M. Study on the region-specific expression of epididymis mRNA in the rams. PLoS One 2021, 16, e0245933. [CrossRef]
- Thimon, V.; Koukoui, O.; Calvo, E.; Sullivan, R. Region-specific gene expression profiling along the human epididymis. Molecular human reproduction 2007, 13, 691–704. [CrossRef]
- Matika, O.; Riggio, V.; Anselme-Moizan, M.; Law, A.S.; Pong-Wong, R.; Archibald, A.L.; Bishop, S.C. Genome-wide association reveals QTL for growth, bone and in vivo carcass traits as assessed by computed tomography in Scottish Blackface lambs. Genetics Selection Evolution 2016, 48, 11. [CrossRef]
- Zhang, L.; Zhang, T.; Yang, Z.; Cai, C.; Hao, S.; Yang, L. Expression of nuclear factor kappa B in ovine maternal inguinal lymph nodes during early pregnancy. BMC Veterinary Research 2022, 18, 266. [CrossRef]
- Kravitz, A.; Liao, M.; Morota, G.; Tyler, R.; Cockrum, R.; Manohar, B.M.; Ronald, B.S.M.; Collins, M.T.; Sriranganathan, N. Retrospective Single Nucleotide Polymorphism Analysis of Host Resistance and Susceptibility to Ovine Johne’s Disease Using Restored FFPE DNA. International Journal of Molecular Sciences 2024, 25, 7748. [CrossRef]
- Kasimanickam, R.; Ferreira, J.C.P.; Kastelic, J.; Kasimanickam, V. Application of genomic selection in beef cattle disease prevention. Animals 2025, 15, 277. [CrossRef]
Figure 1.
Venn diagrams showing shared and unique numbers of SNPs (A) and indels (B) between Royal White and White Dorper sheep.
Figure 1.
Venn diagrams showing shared and unique numbers of SNPs (A) and indels (B) between Royal White and White Dorper sheep.
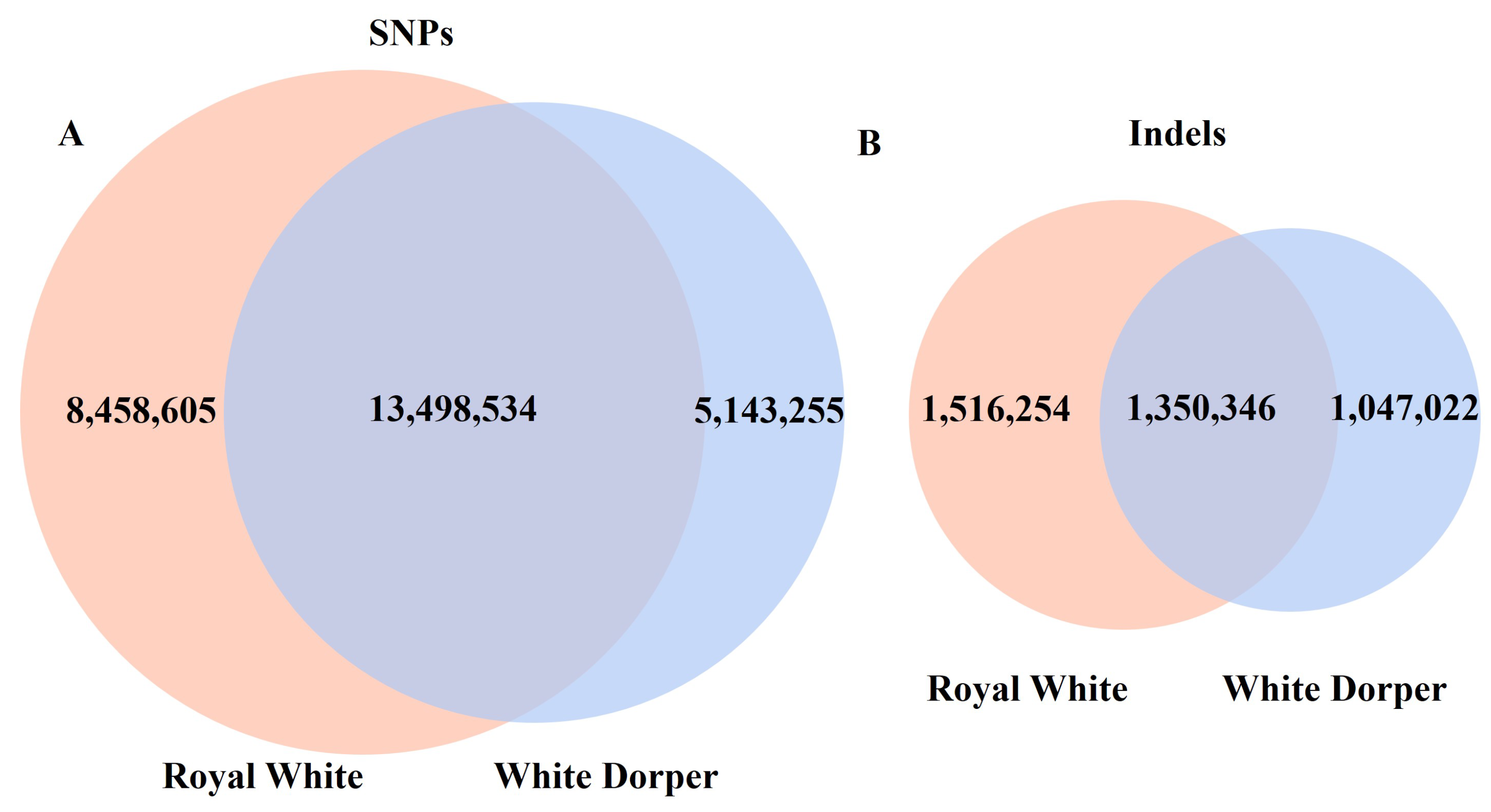
Figure 2.
Indel size distribution in Royal White and White Dorper sheep. Negative values represent deletions, and positive values represent insertions.
Figure 2.
Indel size distribution in Royal White and White Dorper sheep. Negative values represent deletions, and positive values represent insertions.
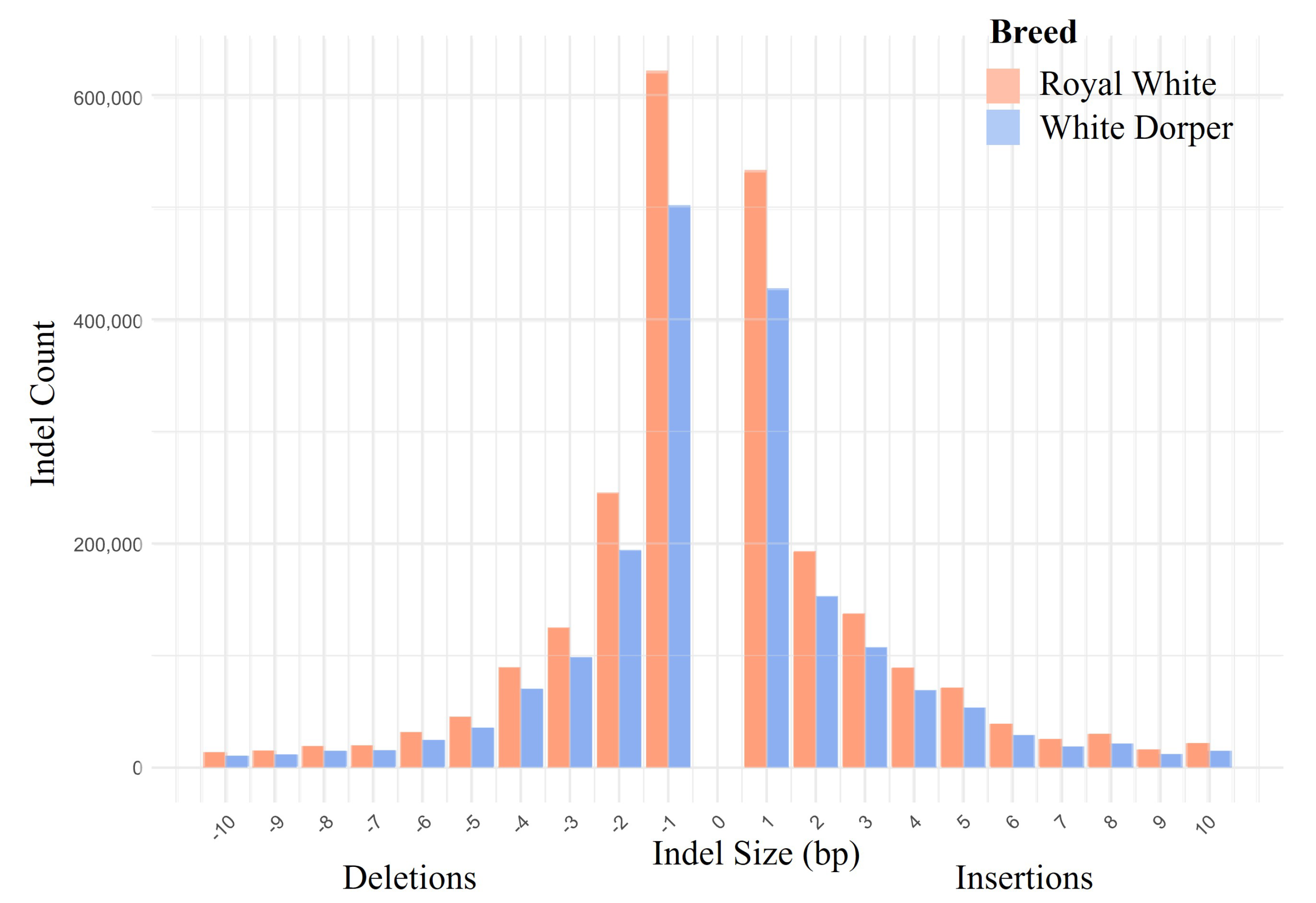
Figure 5.
Principal component analysis (PCA) and discriminant analysis of principal components (DAPC) illustrating population structure between Royal White and White Dorper sheep. (A) PCA plot showing partial separation between the two breeds based on the first two principal components. (B) DAPC density plot showing clear differentiation along the first discriminant function, indicating breed-specific genetic structure.
Figure 5.
Principal component analysis (PCA) and discriminant analysis of principal components (DAPC) illustrating population structure between Royal White and White Dorper sheep. (A) PCA plot showing partial separation between the two breeds based on the first two principal components. (B) DAPC density plot showing clear differentiation along the first discriminant function, indicating breed-specific genetic structure.
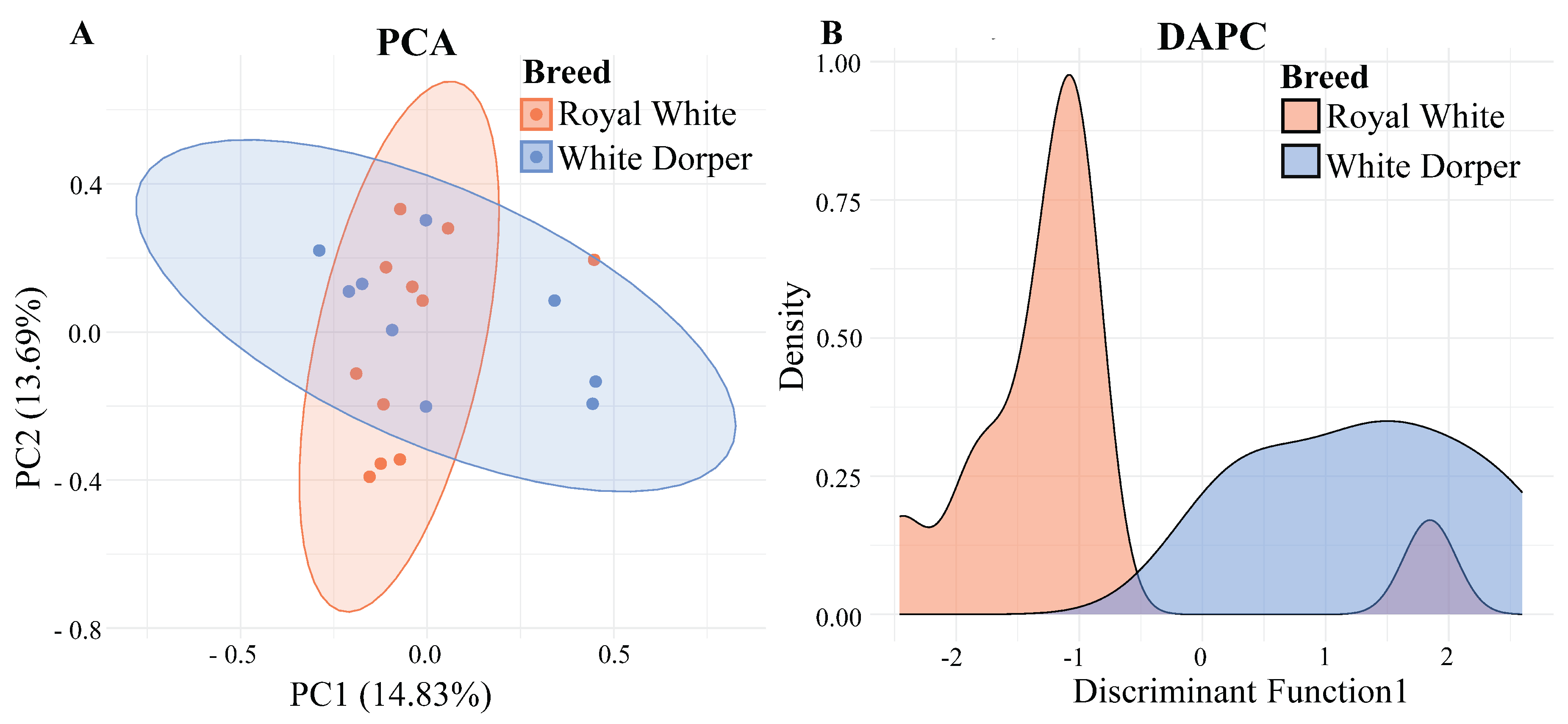
Table 1.
Summary of sequencing metrics including raw reads, cleaned reads, cleaned read retention, mapped read rate, and average depth for Royal White and White Dorper sheep.
Table 1.
Summary of sequencing metrics including raw reads, cleaned reads, cleaned read retention, mapped read rate, and average depth for Royal White and White Dorper sheep.
| Breed | Sample | Raw Reads | Cleaned Reads | Cleaned Reads Retained | Mapped Read Rate | Average Depth |
|---|---|---|---|---|---|---|
| Royal White | RW1 | 66,685,965 | 63,899,137 | 95.82% | 99.93% | 6.53× |
| RW2 | 68,121,792 | 65,611,571 | 96.32% | 99.93% | 6.66× | |
| RW3 | 90,611,600 | 87,965,250 | 97.08% | 99.95% | 8.73× | |
| RW4 | 75,592,210 | 71,813,373 | 95.00% | 99.93% | 7.19× | |
| RW5 | 71,073,803 | 68,690,719 | 96.65% | 99.94% | 7.09× | |
| RW6 | 57,832,096 | 53,349,935 | 92.25% | 99.92% | 5.34× | |
| RW7 | 75,326,235 | 70,831,741 | 94.03% | 99.95% | 6.89× | |
| RW8 | 111,436,645 | 105,628,786 | 94.79% | 99.93% | 10.00× | |
| RW9 | 73,792,997 | 68,491,200 | 92.82% | 99.93% | 6.34× | |
| RW10 | 69,186,458 | 65,441,733 | 94.59% | 99.93% | 6.44× | |
| RW11 | 83,568,451 | 79,745,350 | 95.43% | 99.92% | 7.89× | |
| White Dorper | WD1 | 81,859,706 | 76,544,352 | 93.51% | 99.94% | 7.21× |
| WD2 | 75,404,617 | 69,620,505 | 92.32% | 99.94% | 6.52× | |
| WD3 | 65,262,440 | 60,110,417 | 92.11% | 99.95% | 5.66× | |
| WD4 | 63,321,027 | 54,063,159 | 85.38% | 99.91% | 5.19× | |
| WD5 | 50,539,051 | 46,329,622 | 91.67% | 99.90% | 5.01× | |
| WD6 | 71,909,006 | 68,680,645 | 95.51% | 99.94% | 6.86× | |
| WD7 | 78,854,393 | 75,289,750 | 95.48% | 99.93% | 7.41× | |
| WD8 | 62,284,501 | 59,082,446 | 94.86% | 99.92% | 5.99× | |
| WD9 | 75,691,236 | 72,451,313 | 95.72% | 99.93% | 7.11× |
Table 2.
Summary of filtered SNP and indel counts, Ts/Tv ratios, Het/Hom ratios, and known vs. novel 1 variant classification in Royal White and White Dorper breeds.
Table 2.
Summary of filtered SNP and indel counts, Ts/Tv ratios, Het/Hom ratios, and known vs. novel 1 variant classification in Royal White and White Dorper breeds.
| Metric | Royal White | White Dorper |
|---|---|---|
| SNP | 21,957,139 | 18,641,789 |
| Ts/Tv Ratio | 2.30 | 2.16 |
| Het/Hom (SNP) | 0.999 | 0.998 |
| Known SNP (%) | 16,958,892 (77.24%) | 14,460,461 (77.57%) |
| Novel SNP (%) | 4,998,247 (22.76%) | 4,181,328 (22.43%) |
| Indels | 2,866,600 | 2,397,368 |
| Het/Hom (Indels) | 0.998 | 0.992 |
| Known Indels (%) | 2,240,722 (78.17%) | 1,873,106 (78.13%) |
| Novel Indels (%) | 625,878 (21.83%) | 524,262 (21.87%) |
1 Known and novel classifications are based on comparison with the Ensembl Ovis aries Rambouillet variation database (release 113).
Table 3.
Summary of SNP functional annotation categories in Royal White and White Dorper breeds based on SnpEff.
Table 3.
Summary of SNP functional annotation categories in Royal White and White Dorper breeds based on SnpEff.
| Variant Type | Royal White SNP | White Dorper SNP |
|---|---|---|
| 3’ UTR variant | 335,924 | 286,072 |
| 5’ UTR premature start codon gain | 24,360 | 20,620 |
| 5’ UTR variant | 144,911 | 122,548 |
| Downstream gene variant | 2,330,242 | 1,975,745 |
| Initiator codon variant | 47 | 44 |
| Intergenic region | 11,967,899 | 10,138,113 |
| Intragenic variant | 6,681 | 6,640 |
| Intron variant | 27,465,429 | 23,392,133 |
| Missense variant | 142,906 | 124,264 |
| Non-coding transcript exon variant | 166,581 | 143,434 |
| Splice acceptor variant | 970 | 897 |
| Splice donor variant | 974 | 981 |
| Splice region variant | 42,203 | 36,165 |
| Start lost | 418 | 295 |
| Start retained variant | 1 | 0 |
| Stop gained | 3,525 | 3,886 |
| Stop lost | 304 | 237 |
| Stop retained variant | 185 | 149 |
| Synonymous variant | 195,280 | 165,971 |
| Upstream gene variant | 2,308,159 | 1,951,646 |
Table 5.
Selective sweep regions in Royal White sheep with associated genes and QTL traits.
| Genes1 | Chr | FST2 | SNP IDs3 | QTL Traits4 | Category |
|---|---|---|---|---|---|
| GRM5 | 21 | 0.17 | rs424837012 | Vocalization during arena test | Behavior |
| MAGI2 | 4 | 0.18 | rs429561404 | Locomotion during arena test | Behavior |
| GRM5 | 21 | 0.39 | rs424244818 | Locomotion during isolation box test | Behavior |
| JADE2 | 5 | 0.19 | rs413619557 | Body weight (body weight at 6 months) | Growth |
| ALDH5A1 | 20 | 0.11 | rs421181203 | Mycobacterium avium subsp. paratuberculosis susceptibility (infection status and antibody titer) | Health |
| TGFB2 | 12 | 0.11 | rs160759291 | Gastrointestinal nematode resistance (Haemonchus contortus) | Health |
| TGFB2 | 12 | 0.11 | rs162057314 | Gastrointestinal nematode resistance (Haemonchus contortus) | Health |
| TOX2 | 13 | 0.13 | rs423531735 | Fecal egg count (Haemonchus contortus FEC2) | Health |
| HERC6 | 6 | 0.16 | rs424266480 | Fecal egg count | Health |
| NRXN1 | 3 | 0.24 | rs409057468 | Red blood cell distribution width | Health |
| PARP8 | 16 | 0.17 | rs416975775 | Meat omega-6 to omega-3 fatty acid ratio | Meat |
| NIN | 7 | 0.12 | rs410734119 | Milk yield | Milk |
| NRXN1 | 3 | 0.30 | rs429232758 | Fiber diameter coefficient of variance | Wool |
1 Genes were selected because they contain or overlap SNPs, identified in the sheep QTL database, that are located within selective sweep regions. 2 FST values were calculated using VCFtools in 50 kb sliding windows; the top 10% high-differentiation windows were used to identify candidate regions. 3 SNPs were identified in our dataset and matched to the Ensembl Ovis aries variation database (release 113). 4 QTL trait associations were retrieved from the sheep QTL Database (release 55; file date: 2024-12-23) by mapping the identified SNPs.
Table 6.
Selective sweep regions in White Dorper sheep with associated genes and QTL traits.1–4
| Genes1 | Chr | FST2 | SNP IDs3 | QTL Traits4 | Category |
|---|---|---|---|---|---|
| PLXDC2 | 13 | 0.14 | rs401963094 | Body weight (body weight at 9 months) | Growth |
| COLGALT2 | 12 | 0.14 | rs402132699 | Average daily gain (daily weight gain after nematode challenge) | Growth |
| HYDIN | 14 | 0.29 | rs410323459 | Body weight (body weight at 8 months) | Growth |
| LAMC1 | 12 | 0.13 | rs596561468 | Gastrointestinal nematode resistance (Haemonchus contortus resistance) | Health |
| COLGALT2 | 12 | 0.14 | rs402132699 | Fecal egg count | Health |
| COLGALT2 | 12 | 0.14 | rs402132699 | Fecal egg count (fecal egg count after nematode challenge) | Health |
| COLGALT2 | 12 | 0.14 | rs402132699 | Hematocrit (packed cell volume after nematode challenge) | Health |
| TRIM14 | 2 | 0.18 | rs422296454 | Change in hematocrit (packed cell volume change) | Health |
| EPHA5 | 6 | 0.27 | rs426828157 | Fecal egg count | Health |
| ADD2 | 3 | 0.14 | rs417859328 | Dressing percentage | Meat |
| TENM2 | 5 | 0.11 | rs409487914 | Milk fat yield (180-day milk fat yield) | Milk |
| BUD23 | 24 | 0.18 | rs430795622 | Milk fat yield (180-day milk fat yield) | Milk |
| SCN8A | 3 | 0.40 | rs419496265 | Milk fat percentage | Milk |
| LCN8 | 3 | 0.12 | rs415039972 | Horn number | Morphology |
| NFKB1 | 6 | 0.17 | rs404225841 | Bone area | Morphology |
| STPG3 | 3 | 0.16 | rs430682724 | Offspring number (litter size) | Reproduction |
| DYNC2H1 | 15 | 0.24 | rs413723884 | Offspring number (total number of lambs across first four parities) | Reproduction |
1 Genes were selected because they contain or overlap SNPs identified in the sheep QTL database that are located within selective sweep regions. 2 FST values were calculated using VCFtools in 50 kb sliding windows; the top 10% high-differentiation windows were used to identify candidate regions. 3 SNPs were identified in our dataset and matched to the Ensembl Ovis aries variation database (release 113). 4 QTL trait associations were retrieved from the sheep QTL Database (release 55; file date: 2024-12-23) by mapping the identified SNPs.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



